Reprodução integral do texto de Doença de Machado-Joseph - Hospital de Clínicas de Porto Alegre
O que é a Doença de Machado-Joseph?
A Doença de Machado-Joseph (DMJ) é uma doença crônica que afeta estruturas neurológicas responsáveis principalmente pela coordenação dos movimentos e pelo equilíbrio. Tem um início sutil, em algum momento a vida adulta, e progride de forma gradual, afetando principalmente o caminhar, produzindo oscilações e desvios para os lados e, com o passar do tempo, até mesmo quedas.
A DMJ é uma doença genética. Ela é herdada de modo autossômico dominante, o que quer dizer que todo(a) doente a herdou de um de seus genitores; e que os(as) filhos(as) de um doente poderão também apresentar a condição.
Como são os seus sintomas?
O início da DMJ quase sempre se manifesta por desequilíbrio para caminhar. Isso pode acontecer nas mais variadas idades: houve pacientes que começaram a apresentar a condição aos 10 anos, e houve outros que a apresentaram aos 72 anos. Uma idade que se pode tomar como a média de início, entretanto, seria a dos 32 anos de idade.
Esse desequilíbrio é chamado pelos médicos de "ataxia", e é progressivo. Mas a ataxia não envolve somente o caminhar. Com o passar do tempo, a ataxia pode atingir também a fala, que se torna mal articulada. Em muitos pacientes, esse vai se tornar seu principal problema: a dificuldade de comunicação. Embora intelectualmente normais, essas pessoas não conseguirão se expressar bem através da fala.
A ataxia também pode, com o tempo, atingir os movimentos finos das mãos: os doentes terão dificuldade em escrever, em usar seus utensílios, etc.
Um outro grupo de sintomas importantes são as manifestações oculares. Entre essas manifestações, poderá haver dificuldade de o movimento dos dois olhos ser bem coordenado. Com isso, os dois olhos não fixarão exatamente a mesma imagem cada um. Cada olho poderá fixar pontos diferentes no seu campo de visão. É isso que provoca mais uma dificuldade importante: a visão dupla. A visão dupla é mais aparente quando o paciente olha para longe. Ela é facilmente corrigida se o paciente fechar um dos olhos.
Outros sintomas também podem aparecer, e são às vezes mais perceptíveis para o médico do que para o doente, pois não provocam muita limitação. Em alguns doentes, poderá aparecer uma rigidez que torna os movimentos mais lentos e difíceis de serem executados. Em outros doentes, poderão aparecer uma redução da massa muscular e algum formigamento nos pés.
A DMJ NÃO CAUSA deterioração mental: os pacientes mantêm-se lúcidos, inteligentes e com a memória normal. Entretanto, como se trata de uma condição muito limitante, muitos sofrerão de depressão e de isolamento social. Por isso, podem ser comuns as dificuldades de conciliar o sono, a tristeza e alguma irritabilidade.
Como ela é herdada?
A DMJ é uma doença de herança autossômica dominante. Isso quer dizer que ela é uma "doença vertical", que aparece em todas as gerações de uma família, até onde as pessoas podem se recordar. O diagrama a seguir apresenta uma possível árvore genealógica de uma família com essa doença. Os quadrados simbolizam homens, as bolas simbolizam mulheres. As bolas ou quadrados escuros simbolizam os doentes e as brancas, os sadios. Cada linha horizontal é uma geração. As linhas verticais unem os pais e seus filhos. Imaginem então que esses desenhos simbolizam avós, pais, filhos e netos:
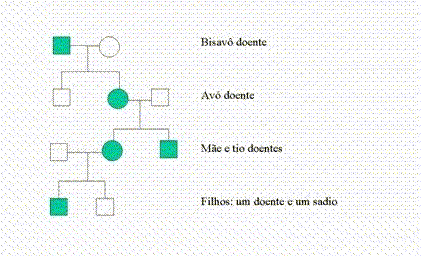
Pode-se perceber que existem filhos de doentes que não herdam a doença (como o irmão da avó, na figura), enquanto outros a herdam. Como isso acontece?
Todos nós herdamos dois genes para cada função corporal. Um gene nós herdamos da mãe e o outro gene, nós herdamos do pai.
O gene que, quando alterado, provoca a doença de Machado-Joseph, é chamado de MJD1. Como nós herdamos dois genes, todos nós temos duas cópias do gene MJD1 - tanto as pessoas sadias, como as doentes. Entretanto, para que nós não tenhamos a doença, as duas cópias do gene MJD1 (tanto a herdada do pai, como a herdada da mãe) devem ser normais. Se uma estiver alterada, mais cedo ou mais tarde ela vai provocar o início da doença. Então, vejamos agora como fica a herança da mesma família mostrada acima, agora com a anotação dos genes de cada pessoa. O gene MJD1 normal, nós vamos chamar de "N"; o gene alterado, nós vamos chamar de "A". Você pode notar que todo mundo, nessa família, vai ter dois genes anotados embaixo de si.
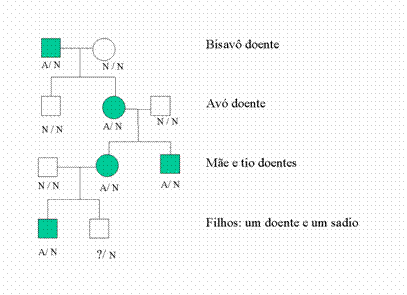
Agora podemos compreender porque um filho de uma pessoa afetada pode não herdar a doença. Observe o irmão da avó: ele não é doente, porque seus dois genes são N. Ele herdou um gene normal de sua mãe sadia (a bisavó da família) e, por sorte, herdou também um gene normal de seu pai doente (o bisavô da família).
Isso acontece porque as pessoas doentes têm dois genes: um gene será alterado (A) e outro gene será quase sempre normal (N). As pessoas doentes transmitem apenas um desses dois genes para cada um de seus(suas) filhos(as). Qualquer um dos dois genes pode ser transmitido. Isso quer dizer que um(a) filho(a) poderá tanto receber o gene N - e nunca apresentar a doença, nem a transmitir para os seus futuros filhos - como poderá receber o gene A, com as mesmas chances. É por isso que os médicos dizem que o risco de um(a) filho(a) herdar a doença de seu pai ou sua mãe doente é de 50%.
Esse tipo de herança é chamado de herança autossômica dominante. Ela é característica da DMJ, bem como de uma série de outras ataxias semelhantes, porém causadas por outros genes. Como o início da doença acontece, em geral, depois da idade reprodutiva, a maioria das pessoas portadoras da DMJ acaba tendo filhos antes de saber que terá a doença.
Como se diagnostica a DMJ?
Foi somente em 1994 que se descobriu qual era exatamente o gene responsável pela doença. Esse gene - o MJD1 do qual já falamos - está localizado dentro do cromossomo 14. A sua função ainda permanece desconhecida. Apesar de não sabermos exatamente para quê esse gene serve, sabemos que ele apresenta uma seqüência repetitiva de moléculas que varia de tamanho. Essas moléculas que se repetem são representadas pelas letras CAG. Uma seqüência CAG, no código genético, codifica o aminoácido glutamina. Os genes normais (N) contêm entre 12 e 37 repetições da seqüência CAG. Os genes alterados (A) contêm mais de 56 repetições CAG.
Essa descoberta tornou possível a realização de um exame molecular, a partir de uma coleta de sangue do paciente. Esse exame "mede" o tamanho das seqüências CAG dos dois genes de cada pessoa. Se uma das seqüências for maior de 56 repetições, então o exame laboratorial identifica a presença de uma mutação. Nós já sabemos que basta um gene alterado - uma mutação - para causar a doença.
A técnica básica desse exame é denominada de PCR (do inglês "polymerase chain reaction"), seguida do acréscimo de uma sonda de DNA que marca as CAGs. O material genético depois passa por uma eletroforese, o que separa os pedaços de DNA de acordo com seu tamanho. Isso possibilita identificar pedaços pequenos (com poucas CAGs - os que se encontram nos genes normais) e pedaços grandes (com muitas CAGs - os que se encontram nos genes mutantes ou alterados).
O que causa a DMJ?
Se você leu a seção acima, pode compreender o que sabemos sobre a causa da DMJ. Se toda a pessoa doente tem um gene com uma seqüência CAG grande, maior do que 56 repetições, então é isso que causa a doença.
Mas não sabemos ainda exatamente porque isso causa a doença.
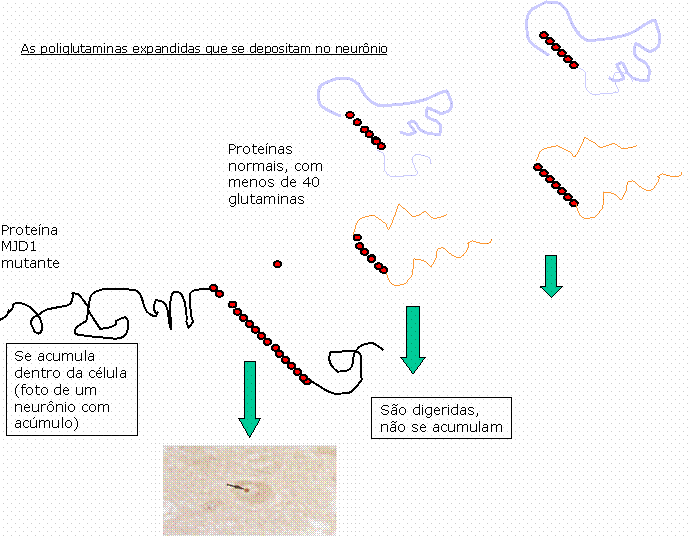
O que sabemos é que a seqüência CAG do gene produz, por sua vez, uma proteína na qual haverá repetidos aminoácidos glutamina. Se os genes de uma pessoa contêm, por exemplo, cada um, 15 e 70 repetições CAG, isso quer dizer que uma parte das proteínas por eles produzida terá 15 glutaminas, e outra parte terá 70 glutaminas. Sabemos que, quando uma proteína contém mais de 40 glutaminas, essa proteína não é mais digerida dentro da célula, e passa a se acumular dentro das estruturas celulares. Essa é provavelmente a causa da doença. Nossos neurônios não foram feitos para acumular essas "poliglutaminas". É possível que o seu acúmulo provoque a disfunção dos neurônios e, mesmo, sua morte.
As poliglutaminas expandidas que se depositam no neurônio
O que sabemos da origem histórica da DMJ?
A DMJ foi primeiro descrita em 1972 por dois grupos de investigadores norte-americanos. A primeira família descrita descendia de William Machado, um habitante da ilha de São Miguel, no arquipélago dos Açores. A segunda família também descendia de açorianos provenientes dessa mesma ilha. Essas duas famílias norte-americanas moravam, na ocasião, na costa leste dos Estados Unidos. Em 1976, uma terceira família foi identificada, dessa vez na costa oeste daquele país. Essa família também era de origem açoriana (porém da ilha de Flores) e se chamava "Joseph". A partir desse ano, a comunidade de investigadores clínicos concluiu que as três famílias deveriam ter a mesma doença, que passou a ser chamada com o nome das primeiras famílias reconhecidas: "Machado-Joseph".
Como se reconheceu que em todas havia uma ancestralidade açoriana, um grupo de investigadores partiu para aquelas ilhas. Reconheceu-se, então, que entre os açorianos essa doença era muito mais freqüente do que se esperava.
Hoje em dia, sabemos há doentes com DMJ nas mais variadas populações: entre portugueses, franceses, alemães, norte americanos, negros norte e sul-americanos, entre japoneses, chineses, indianos e até mesmo entre aborígenes australianos. Existem evidências de que muitos desses doentes tenham herdado essa condição de longínquos antepassados portugueses. Você deve se lembrar de como os portugueses eram importantes e dominavam o comércio marítimo há 400-500 anos atrás. Supõe-se que foi justamente através das navegações daquela época, que a DMJ se difundiu.
No Rio Grande do Sul, a história foi mais ou menos semelhante. Nosso Estado praticamente não tinha população até a vinda dos açorianos, a partir de 1750. Com essa imigração, nossa população surgiu e se estabeleceu. É bem possível que alguns dos imigrantes daquela época fossem portadores do gene alterado, e que o tenham transmitido aos seus descendentes até as gerações atuais. Sabemos, por exames moleculares, que os doentes gaúchos herdaram o gene que se originou da ilha de Flores, no arquipélago açoriano.
O que se pode fazer para melhorar seus sintomas?
Embora não exista um tratamento que interrompa o curso da doença, ou que previna a doença entre as pessoas ainda sem sintomas, muitos cuidados podem ser tomados para melhorar a qualidade de vida dos doentes.
Os principais cuidados são a fisioterapia motora e a fonoaudiologia. Ambas auxiliam na preservação das funções motoras. Ensinam aos pacientes como lidar com situações tais como o engasgo e a marcha desequilibrada.
Eventualmente, pode ser necessário o uso de um medicamento para diminuir algum sintoma. Esse parece ser o caso quando o paciente apresenta muita rigidez. Alguns remédios diminuem a rigidez, mas seu uso deve ser bem ponderado, pois efeitos colaterais podem aparecer. O especialista habilitado na sua prescrição é o neurologista.
Em outros casos, mais comuns, o uso de uma medicação anti-depressiva está indicado e pode trazer grande alívio dos sintomas. Já comentamos que a DMJ traz muita incapacitação funcional: o doente deixa paulatinamente de caminhar, de se comunicar e de realizar suas tarefas. Isso traz um enorme sentimento de perda e de tristeza. Esse sentimento - essa depressão - pode, por sua vez, vir a piorar os sintomas motores, produzindo ainda mais limitação. Por isso, é possível que, em muitos casos, o uso judicioso de um anti-depressivo melhore a qualidade de vida do doente e mesmo de sua família.
O que se pode fazer para ajudar as pessoas que estão em risco de herdarem a DMJ?
As pessoas de uma família que tem o diagnóstico de DMJ, podem querer fazer o teste para saber se herdaram a doença antes mesmo de manifestarem os sintomas. Este teste, realizado em pessoas assintomáticas é chamado de "Teste Preditivo para a DMJ".
Este teste é freqüentemente solicitado pelos interessados, para auxiliar suas decisões relacionadas aos seus planos futuros no que diz respeito à família, ao trabalho, etc. Outras pessoas fazem o teste apenas por "necessitar saber".
Os laboratórios costumam realizar esse teste nos indivíduos que tenham 50% de risco de virem a apresentar a condição. É o caso do segundo filho (da última linha) apresentado na Figura 2. Ele é sadio e jovem, e não se sabe ainda se herdou ou não a mutação de sua mãe.
O Teste Preditivo para DMJ compreende uma pré-avaliação onde serão revisados os motivos que levam à decisão de realizá-lo, e o possível impacto de um resultado positivo ou negativo. Este teste é confidencial e sigiloso. O seu resultado somente é dado para o indivíduo que o realizou. Não fica no arquivo hospitalar dessa pessoa.
Como não se conhece ainda uma cura ou um tratamento preventivo para a DMJ, é importante que se compreenda que esse teste preditivo não trará conseqüências "médicas" - quer dizer, medicações ou outros tratamentos.
Uma vez que a motivação para se realizar esse teste é de foro íntimo e individual, e que ao mesmo tempo não há um tratamento disponível, esse teste preditivo não deve ser realizado em crianças e adolescentes.
Onde você pode encontrar mais informações e cuidados?
1) Você pode procurar atendimento no nosso ambulatório, ou seja, no:
Programa de Doenças Neurogenéticas
Ambulatório de Ataxias
Serviço de Genética Médica
Hospital de Clínicas de Porto Alegre
Rua Ramiro Barcelos, 2350
90.035-903 Porto Alegre Brasil
Fones (51) 33168309 e 33168423
Fax (51) 33168010
2) Há inúmeras páginas da Internet nas quais você pode obter mais informações. Abaixo, nos listamos os mais conhecidos:
a) Associação Lusa: www.lusaweb.com/
b) Universidade do Porto: webhome.idirect.com/~albri/
c) National Institute of Neurological Disorders and Stroke, Maryland, EUA: www.ninds.nih.gov
d) International Joseph Disease Foundation, California, EUA: www.ijdf.net
e) National Organization for Rare Disorders (NORD): www.rarediseases.org
f) National Ataxia Foundation, do Minessota, EUA: www.ataxia.org
g) International Network of Ataxia Friends (INTERNAF): www.wemove.org
SCA3 - Doença Machado-Joseph (extraído de "Living With Ataxia")
A ataxia ou doença de Machado-Joseph (MJD), também conhecida como SCA3 - ataxia espinocerebelar tipo 3, é uma forma de ataxia hereditária autossômica dominante (cromossomo 14). O nome Machado-Joseph provém dos nomes das duas primeiras famílias descritas com esta condição, em 1972. Ambas as famílias eram originárias das ilhas portuguesas de Açores. Inicialmente pensou tratar-se de uma enfermidade muito rara somente encontrada em certos grupos étnicos isolados, mas a pesquisa genética revelou que a SCA3 é de fato mais comum do que a SCA1. O gene responsável pela SCA3 foi identificado por uma equipe de pesquisadores do Japão, em 1993. Assim como a SCA1, um teste genético pode diagnosticar com precisão a presença ou a ausência do gene alterado que causa SCA3.
Os sintomas podem ser mais amplos do que os da SCA1. Como na SCA1, a enfermidade normalmente começa na idade adulta e progride através dos anos. Já foram constatados casos com início na adolescência e outros até com a idade de 70 anos. Como em todas as formas de ataxia, o primeiro sintoma geralmente é a falta de equilíbrio, seguido depois pela falta de coordenação nas mãos e dificuldades na fala. Alguns indivíduos notam visão dupla. Para o médico, a limitação de movimentos dos olhos, movimentos oculares anormalmente lentos ou o olhar fixo pode ser um sinal de que se trata de SCA3.
Com a evolução, sintomas neurológicos adicionais como espasticidade, rigidez, perda de volume e força muscular e lentidão de movimentos são comuns. Embora ainda não exista tratamento para essa enfermidade, sintomas como fadiga, depressão, distúrbios do sono, dores e tremores, presentes em alguns indivíduos afetados, podem melhorar com medicação adequada.
