GENERALITES SUR L'OZONE
I. Généralités sur l'ozone
Dans l'antiquité déjà,
on connaissait l'ozone par son odeur caractéristique qui survient
après les éclairs, ainsi que le relatent les épopées
d'Homère : l'Iliade et l'Odyssée dans lesquelles l'on qualifie
cette odeur de « sulfureuse ». Ce n'est que depuis deux siècles
que le Hollandais Van Marum (Leipzig, 1785) a décelé la réduction
de volume accompagnant l'apparition de cette odeur lors de décharges
électriques produites dans l'air. En 1801, W. Cruicksank observa
une « odeur semblable à celle du chlore » durant l'électrolyse
d'acide sulfurique. L'ozone n'a été en fait découverte
qu'en 1840 par le Suisse M. F. C. Schönbein(Schonbein
1840) qui la dénomma ainsi en se référant à
la racine grecque Ozeïn : exhaler une odeur, sentir. Les recherches
seront facilitées en 1857 par la construction par W. Von Siemens
du premier ozoneur et dont le principe, basé sur un tube à
décharge, est encore utilisé actuellement. J. L. Soret fut
le premier, en 1865, grâce à une détermination indirecte
de la densité, à démontrer la composition chimique
de l'ozone conduisant à la formule O3. « ...l'hypothèse
la plus logique pour expliquer cet ensemble de faits, consiste à
admettre que l'ozone est une modification allotropique de l'oxygène...
» (Otto 1897). La découverte des
propriétés bactéricides de l'ozone par Ohlmüller
en 1890, a été à l'origine de sa production industrielle.
Ainsi, à la suite des expériences de De Méritens réalisées
à l'usine de Saint-Maur, la première centrale d'ozonation
a été construite sur le site de Bon Voyage. Cette usine,
dès 1907, a permis le traitement de 22500 m3 / jour d'eau
captée de la Vésubie afin d'alimenter la ville de Nice. Malgré
ses nombreux avantages, les problèmes de conception des ozoneurs
ont conduit les industries de traitement sinon à préférer
le chlore du moins à l'allier à l'ozone. Il faut noter à
titre anecdotique que de l'ozone pur a été produit en 1922
par E. H. Riesenfeld et G. M. Schwab. Depuis la dernière guerre
lors de laquelle l'ozone a été utilisé dans le développement
des fusées son développement a connu un véritable
essor. Ainsi en 1960 a été développé un procédé
de stockage de l'ozone dans certains fréons(Mahieux
1960).
Ce sont surtout les travaux
de Criegee(Criegee 1953; Criegee
1957) qui à partir de 1953 ont fait avancer nos connaissances
dans le domaine de la chimie de l'ozone. L'ozone est maintenant utilisé
dans l'agriculture, l'alimentation, l'industrie chimique et surtout dans
le domaine du retraitement des eaux. Le développement de l'écologie
et des nouvelles technologies ont initié de nombreuses applications
de l'ozone dans, des domaines aussi divers que, le blanchiment et la délignification
de la pâte à papier, l'industrie textile, les traitements
sanitaires et médicaux, le traitement de l'air, l'industrie des
cosmétiques, l'industrie laitière, etc...
En outre, la découverte
à partir de 1965 de la disparition de la couche d'ozone a fait prendre
conscience aux scientifiques par le biais de l'opinion mondiale de l'importance
de l'ozone et le développement des programmes d'étude de
la chimie de l'ozone en résulte. Ces travaux ont été
consacrés en 1995 par l'attribution du prix Nobel de chimie à
Paul Crutzen, Mario Molina, et F. Sherwood Roland pour leurs travaux sur
la disparition de la couche d'ozone.
2. Données physico-chimique sur l'ozone
L'ozone est un composé
naturel présent dans toute l'atmosphère à très
faible concentration. Dans la stratosphère, il joue le rôle
d'un filtre protecteur en absorbant les radiations ultra-violettes de longueur
d'onde comprise entre 200 et 300 nm, le maximum d'absorption se situant
à 254 nm. Cette propriété est d'ailleurs utilisée
pour la détermination de la concentration en ozone dans un gaz.
L'ozone peut être généré
de multiples façons et nous pouvons citer(Nebel
1981) :
-
la thermolyse de l'oxygène
à 2760 °C suivie d'une trempe dans l'oxygène liquide,
-
l'oxydation lente du phosphore,
-
l'irradiation de l'oxygène
par des rayons b,
-
l'électrolyse de solutions
aqueuses de phosphate,
-
l'électrolyse d'acide sulfurique
concentré à basse température,
-
l'irradiation d'oxygène
ou d'air par les rayons ultra-violets,
-
la décharge électrique
dans un flux d'air ou d'oxygène.
A température ordinaire
l'ozone est un gaz instable de couleur bleue lorsqu'il est observé
sous une épaisseur suffisante, d'odeur caractéristique et
pénétrante (décelable à des teneurs de l'ordre
de 0,01 à 0,05 ppm). Il est soluble dans certains solvants organiques
tels que l'acide acétique, l'anhydride acétique, l'acétate
d'éthyle, le trichlorométhane, le tétrachlorométhane,
des dérivés chlorofluorés. Les principales constantes
physiques, thermodynamiques et toxicologiques de l'ozone sont reprises
dans le Tableau II-1.Tableau II-1 : Principales constantes physiques
de l'ozone.

La solubilité de l'ozone
dans l'eau est fonction de la température (il est d'autant plus
soluble que la température est basse), et de la pression partielle
en ozone22, 23 (Figure II-1).
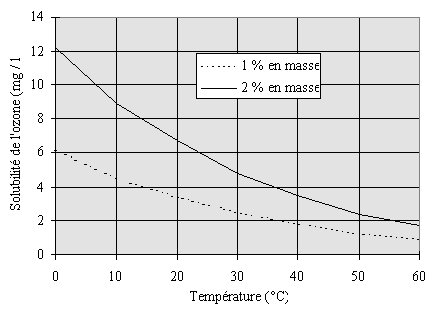
Figure II-1 : Solubilité
de l'ozone dans l'eau vs. température à la pression atmosphérique
à deux concentrations différentes en ozone.
L'ozone se présente sous
la forme d'un triangle isocèle d'angle au sommet égal à
116°45' 65'. Les deux liaisons interatomiques ont pour longueur 1,278
0,003 Å(Manley and Niegowski 1967).
La molécule peut être considérée comme un hybride
de résonance de quatre formes mésomères. L'atome central
des quatre formes mésomères possède un octet entier
d'électrons, les atomes externes des formes III et IV n'ont qu'un
sextet. Ce sont des positions électrophiles.
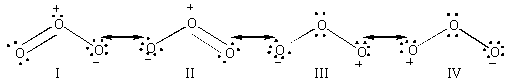
Figure II-2 :
Les quatre formes limites de résonance de l'ozone.
La molécule d'ozone
possède en outre un faible moment dipolaire de 0,53 Debye et ne
présente pas de paramagnétisme appréciable.
II. Réactivité de l'ozone
1. Comportement de l'ozone dans l'eau
En solution aqueuse, l'ozone peut
se décomposer avant de réagir avec le substrat et cette décomposition
conduit à la formation d'espèces plus réactives. Divers
travaux ont permis de démontrer que la décomposition de l'ozone
résultait d'une réaction en chaîne dans laquelle l'ion
hydroxyde joue le rôle d'initiateur. Les espèces radicalaires
formées au cours de la réaction initient la réaction
en chaîne. L'ozone peut donc soit réagir directement avec
un substrat (S) ou au delà d'une valeur critique du pH se décomposer.
Dans ce dernier cas les espèces issues de la décomposition
de l'ozone dans l'eau telles que le radical hydroxyle (°OH) peuvent
devenir les agents oxydants les plus importants en solution. La valeur
critique du pH au delà de laquelle ce second type de réaction
devient prédominant dépend de la vitesse avec laquelle l'ozone
réagit avec le substrat ainsi qu'avec les solutés (en tenant
compte des sous-produits de la réaction qui peuvent accélérer
ou retarder la décomposition de l'ozone) (Figure II-3) (Hoigne
and Bader 1976; Hoigne and Bader 1978).
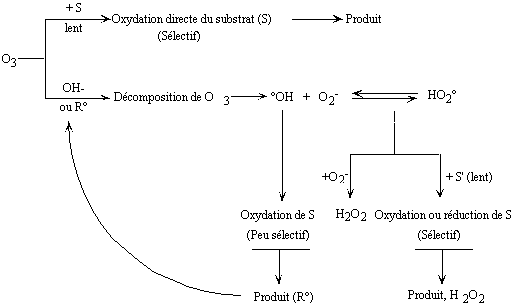
Figure II-3 : Différents
modes d'interaction de l'ozone en solution aqueuse d'après Hoigné
et coll.(Hoigne and Bader 1976; Hoigne
and Bader 1978).
Nous allons successivement
aborder le problème des radicaux générés par
la décomposition de l'ozone dans l'eau pour aborder ensuite la réactivité
de l'ozone moléculaire.
2. Décomposition de l'ozone dans l'eau
a. Mécanisme de décomposition
: formation de radicaux libres en solution aqueuse
La stabilité de l'ozone
dissous dans l'eau dépend fortement du pH, de la lumière
ultraviolette incidente, de la concentration en ozone et de la présence
d'inhibiteurs, de propagateurs ou d'initiateurs de radicaux libres en solution(Tomiyasu,
Fukutomi et al. 1985). En revanche, la température ne semble
jouer qu'un rôle mineur entre 15 °C et 35 °C(Pan,
Chen et al. 1984; Parthasarathy
and Peterson 1990). La cinétique de décomposition de
l'ozone dans l'eau pure peut être formulée selon(Peleg
1976; Staehelin and Hoigné 1982;
Parthasarathy and Peterson 1990)
:
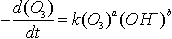
avec a variant de 1 à
2 selon les auteurs et b de 0,36 à 1 selon le pH (se rapproche de
1 à pH acide) et k = 70 M-1s-1
Les équations décrivant
le mécanisme sont les suivantes(Weiss
1935; Staehelin and Hoigné 1982)
:
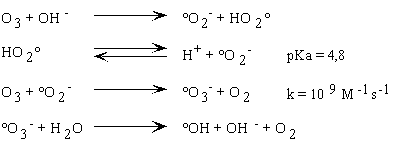
En présence de peroxyde
d'hydrogène sous la forme anionique (HO2-)
et à une concentration supérieure à 10-7
M (pH < 12), l'ozone se décompose beaucoup plus rapidement qu'en
présence d'anion hydroxyde. La cinétique de la réaction
de décomposition peut alors s'écrire selon :
 avec k = 2,8 106 M-1s-1
avec k = 2,8 106 M-1s-1
ce qui conduit aux équations
suivantes :

Le mécanisme de décomposition
de l'ozone a été formalisé par Hoigné, Staehelin
et Bader(Hoigne and Bader 1975; Hoigne
and Bader 1976; Hoigne and Bader 1978;
Hoigne and Bader 1979; Bader
and Hoigné 1981; Staehelin
and Hoigné 1981; Staehelin and
Hoigné 1982; Hoigne and Bader
1983; Hoigne and Bader 1983; Hoigne,
Bader et al. 1985; Staehelin and Hoigné
1985). Ils ont proposé deux processus en absence de substrat
(Figure II-4) et en présence de substrat (Figure II-5).
 Figure
II-4 : Réactions de décomposition de l'ozone dans
l'eau pure (Staehelin and Hoigné
1985).
Figure
II-4 : Réactions de décomposition de l'ozone dans
l'eau pure (Staehelin and Hoigné
1985). 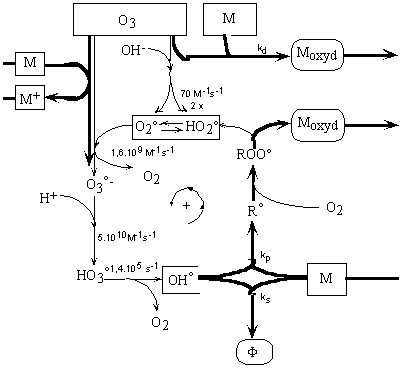
Figure II-5 :
Réactions de l'ozone dans l'eau en présence de solutés
M pouvant réagir avec O3 et interagir avec °OH en
piégeant ou convertissant ce radical en HO2° (Staehelin
and Hoigné 1985).
La réaction entre l'ozone et OH-conduit
à la formation d'un ion superoxyde (O2°-)
et d'un radical hydroperoxyle (HO2°) selon un équilibre
acido-basique de pK = 4,8. La constante cinétique est donc de 2
x 70 M-1s-1.
ii. Les étapes de propagation
La réaction se propage par décomposition
de O3°- et HO3° en radical °OH
qui peut ensuite réagir avec un soluté M. L'ordre de grandeur
de la constante cinétique relative à ce type de réaction
entre le radical hydroxyle et un substrat(Anbar
and Neta 1967; Farhataziz and Ross
1977; Hoigne and Bader 1979; Hoigne
and Bader 1983; Hoigne and Bader 1983;
Hoigne, Bader et al. 1985) est de l'ordre
de 108-1010 M-1s-1. Certains
composés organiques réagissent avec le radical hydroxyle
pour conduire à la formation de radicaux organiques qui après
addition d'une molécule d'O2 éliminent le couple
HO2°-/O2°-.
La constante cinétique avec laquelle O2°-
réagit avec l'ozone est beaucoup plus grande que celle de la réaction
avec les autres solutés présents dans le milieu. Aussi la
conversion du radical peu sélectif °OH vis à vis de l'ozone
en le radical très sélectif O2°-
provoque la réaction en chaîne.
iii. Les étapes de terminaison
Beaucoup de composés organiques peuvent
réagir avec °OH sans pour autant conduire à la formation
du couple HO2°-/O2°-.
Ce type de composés est appelé inhibiteur de radicaux libres.
b. Rôle des inhibiteurs,
initiateurs et promoteurs de radicaux libres
A partir des mécanismes
décrits précédemment, il est aisé de définir
3 types d'évolution possibles des substrats.
i. Les initiateurs de radicaux libres
Ce sont des composés capables d'induire
la formation de l'ion superoxyde O2°-
à partir d'une molécule d'ozone. Nous pouvons citer : OH-,
HO2-, différents
cations métalliques, l'acide glyoxylique, l'acide formique, la lumière
ultraviolette, le peroxyde d'hydrogène. En ce qui concerne les cations
métalliques, de nombreuses études ont été réalisées
afin de déterminer leur réactivité vis à vis
de l'ozone(Hill 1948; Pan,
Chen et al. 1984; Parthasarathy
and Peterson 1990). Il a ainsi pu être mis en évidence
que la présence d'ions de métaux lourds entraînait
une décomposition très rapide de l'ozone. Un classement définissant
l'efficacité a été proposé pour différents
cations métalliques : Co (II) >> Fe (II) > Fe (III) > Mn (II) >
Cu (II). Les mécanismes entrant en jeu sont du type observé
pour le couple OH-
/ O3 conduisant à la formation de OH°. Des études
complémentaires ont montré qu'en présence de métaux
et d'acide acétique l'ozone était stabilisé (Walling
and El-Taliawi 1973). Plusieurs interprétations ont été
formulées pour expliquer cet effet dont la plus classique est d'envisager
la complexation du métal par l'acide.
ii. Les promoteurs de radicaux libres
Ce sont des composés capables de régénérer
l'ion superoxyde O2°-
à partir du radical hydroxyle (Figure II-6). Nous pouvons citer
les composés comportant un groupement aryle, l'acide formique, l'acide
glyoxylique, les alcools primaires ainsi que les ions phosphate.
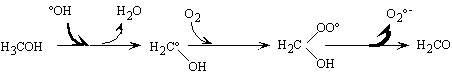
Figure II-6 : Réaction
type d'un initiateur de radicaux libres ; ici le méthanol d'après
Staehelin et coll. (Staehelin and Hoigné
1985).
iii. Les inhibiteurs de radicaux libres
Ce sont des composés capables de réagir
avec le radical hydroxyle sans régénérer l'ion superoxyde
O2°-(Figure
II-7). Parmi ces dérivés nous pouvons citer : les ions carbonate
et bicarbonate, les groupements alkyle, les alcools tertiaires (tel que
l'alcool tertiobutylique ou le tert-butanol).
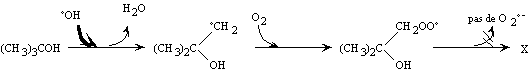
Figure II-7 :
Réaction type d'un inhibiteur de radicaux libres ; ici l'alcool
ter-butylique d'après Staehelin et coll. (Staehelin
and Hoigné 1985).
Des calculs ab initio réalisés
sur la molécule d'ozone ont abouti à la conclusion que cette
molécule peut-être considérée dans son état
fondamental comme un diradical(Goddard,
Dunning et al. 1973; Wadt and Goddard
1975). Cette structure ne permet cependant pas de rendre compte du
caractère électrophile de la molécule. Sur les bases
des structures résonantes décrites précédemment
nous pouvons dire que l'ozone peut agir soit comme un agent d'addition
1,3-dipolaire(Huisgen 1963), comme
un réactif électrophile(Meinwald
1955) ou nucléophile.
III. Différents types de réaction
rencontrés avec l'ozone
1. Réaction de cycloaddition (mécanisme
de Criegee)
Pour rendre compte de la réaction
de cycloaddition, la structure dipolaire de l'ozone doit être envisagée.
Cette réaction peut intervenir au niveau des liaisons insaturées.
La réaction d'addition 1,3-dipolaire peut également concerner
une liaison carbone-hydrogène activée ou non. Une liaison
est dite activée si l'état de transition au cours de l'ozonation
est fortement stabilisé par un groupe donneur d'électrons
tel qu'un groupe hydroxyle, alkoxy ou amino. Les molécules concernées
sont donc les alcools, les éthers, les aldéhydes et les amines.
Les composés ne comportant pas de liaison activée sont les
alcanes et les cycloalcanes.
2. L'attaque électrophile
L'attaque électrophile
peut avoir lieu sur des molécules à forte densité
électronique telles que les composés aromatiques possédant
un substituant électrodonneur et conduisant à la formation
de quinone, et les oléfines qui peuvent être époxydées.
3. Réaction d'autooxydation
Deux mécanismes ont été
proposés pour l'oxydation de liaisons carbone-hydrogène ;
l'un correspond à une addition 1,3-dipolaire(Price
and Tumolo 1964; White and Bailey 1965;
Batterbee and Bailey 1967; Bach,
Su et al. 1994), l'autre à une réaction de type radicalaire
avec initiation par l'ozone d'une autooxydation(Carceller-Fernandez
1940; Schubert and Pease 1956; Schubert
and Pease 1956). Cette réaction radicalaire est une réaction
en chaîne dont l'ozone est le précurseur et dont l'oxygène
est l'oxydant.
4. L'attaque nucléophile
Prévue par la théorie,
l'attaque de l'ozone sous la forme d'un diradical a été observée
lors de l'oxydation de composés possédant un déficit
électronique(Langlais, Reckhow et al. 1985).
D. BIBLIOGRAPHIE
Anbar,
M. and P. Neta (1967). "A compilation
of Specific Bimolecular Rate Constants for the Reactions of Hydrated Electrons,
Hydrogen Atoms and Hydroxyl Radicals with Inorganic and Organic Compounds
in Aqueous Solution." International Journal of Applied Radiation
and Isotopes 18: 493-523.
Bach,
R. D., M.-D. Su, et al. (1994). "Oxidation
of Amines and Sulfides with Hydrogen Peroxide and Alkyl Hydrogen Peroxide.
The Nature of the Oxygen-Transfer Step." Journal of the American
Chemical Society 116: 5379-5391.
Bader,
H. and J. Hoigné (1981). "Determination
of ozone in water by the indigo method." Water Research 15:
449-456.
Bailey, P. S.
(1978). Ozonation in Organic Chemistry. New York, Academic Press.
Bailey, P. S.
(1978). Ozonation in Organic Chemistry. New York, Academic Press.
Batterbee,
J. E. and P. S. Bailey (1967). "Ozonation
of Carbon-Hydrogen Bonds. Anthrone." Journal of Organic Chemistry
32(32): 3899-3903.
Carceller-Fernandez,
C. C. (1940). An. R. Soc. Esp. Fis. Quim. 36: 235-240.
Criegee,
R. (1953). Justus Liebigs Annalen Chemistry 564: 9-20.
Criegee,
R. (1957). "The Course of Ozonation
of Unsaturated Compounds." Record of Chemical Progress 18(2):
110-120.
Farhataziz
and A. B. Ross (1977). "Selected Specific
Rates of Reactions of Transients from Water in Aqueous Solutions. III.
Hydroxyl Radical and Perhydroxyl Radical and their Radical Ions." National
Bureau of Standards, DC; NSRDS-NBS59 59: 1-122.
Forni,
L., D. Bahnemann, et al. (1982). "Mechanism
of the Hydroxide Ion Initiated Decomposition of Ozone in Aqueous Solution."
Journal of Physical Chemistry 86: 255-259.
Goddard,
W. A., T. H. Dunning, et al. (1973). Account of Chemical Research
6: 368-375.
Hill,
G. R. (1948). "Kinetics, Mechanism, and
Activation Energy of the Cobaltous Ion Catalyzed Decomposition of Ozone."
Journal of the American Chemical Society 70: 1306-1307.
Hoigne,
J. and H. Bader (1975). "Ozonation of water : Role of hydroxyl Radicals
as Oxidizing Intermediates." Environemental Science and Technology
109: 4216-4225.
Hoigne,
J. and H. Bader (1976). "The role of hydroxyl
radical reactions in ozonation processes in aqueous solutions." Water
Research 10: 377-386.
Hoigne,
J. and H. Bader (1978). "Ozone initiated
oxidations of solutes in wastewater : a reaction kinetic approach." Progress
in Water Technology 10(5/6): 657-671.
Hoigne,
J. and H. Bader (1979). "Ozonation of
water : selectivity and rate of oxidation of solutes." Ozone : Science
& Engineering 1: 73-85.
Hoigne,
J. and H. Bader (1983). "Rate constants
of reactions of ozone with organic and inorganic compounds in water-I Non-dissociating
organic compounds." Water Research 17: 173-183.
Hoigne,
J. and H. Bader (1983). "Rate of reactions
of ozone with organic and inorganic compounds in water-II Dissociating
organic compounds." Water Research 17: 185-194.
Hoigne,
J., H. Bader, et al. (1985). "Rate constants
of reactions of ozone with organic and inorganic compounds in water-III
Inorganic compounds and radicals." Water Research 19(8):
993-1004.
Huisgen,
R. (1963). Angewandte Chemie, International Edition in English 2:
565-570.
Langlais, B.,
D. A. Reckhow, et al. (1985). Ozone
in Water Treatment : Application and Engineering, LEWIS.
Mahieux,
F. (1960). "Quelques manipulations
de laboratoire avec de l'ozone à l'état liquide et solide."
Bulletin de la Société Chimique de France: 746-749.
Manley,
T. C. and S. J. Niegowski (1967). . Kirk-Othmer Encyclopedia of Chemical
Technology. London, Interscience Publisher. 14: 410-432.
Meinwald,
J. (1955). Chemische Berichte 88: 1889-1920.
Nebel,
C. (1981). . Kirk-Othmer Encyclopedia of Chemical Technology. London,
Interscience Publisher. 16: 683-713.
Otto, M.
(1897). 1 ère thèse. Paris, 1ère thèse.
Pan,
G. Y. Y., C.-L. Chen, et al. (1984). "Studies
on ozone bleaching. I. The effect of pH, temperature, buffer systems and
heavy metal-ions on stability of ozone in aqueous solution." Journal
of Wood Chemistry and Technology 4(3): 367-387.
Parthasarathy,
V. R. and R. C. Peterson (1990). Ozone
bleaching, Part I. The decomposition of ozone in aqueous solution - Influence
of pH, temperature and transition metals on the rate kinetics of ozone
decomposition. Oxygen Delignification, TAPPI.
Pascal,
P. (1960). . Nouveau traité de chimie minérale. Paris.
XIII: 243-312.
Peleg,
M. (1976). "The chemistry of ozone in
the treatment of water." Water Research 10: 361-365.
Price,
C. C. and A. L. Tumolo (1964). "The
course of ozonation of Ethers." Journal of the American Chemical
Society 86: 4691-4694.
Rio,
M. (1962). "Nouveau traité de chimie minérale." Revue
de L'Air Liquide 40: 20-25.
Schonbein,
C. (1840). Hebd. Scanees Acad. Sci. 10: 706-710.
Schubert,
C. C. and R. N. Pease (1956). "The
oxidation of Lower Paraffin Hydrocarbons. I. Room Temperature Reaction
of Methane, Propane, n-Butane and Isobutane with Ozonized Oxygen." Journal
of the American Chemical Society 78: 2044-2048.
Schubert,
C. C. and R. N. Pease (1956). "The
oxidation of Lower Paraffin Hydrocarbons. II. Observations on the Role
of Ozone in the Slow Combustion of Isobutane." Journal of the American
Chemical Society 78: 5553-5556.
Staehelin,
J. and J. Hoigné (1981). Zur chemischen
reaktionskinetik des ozonzerfalls in wasser. Ozone in Water Treatment,
Berlin, IOA.
Staehelin,
J. and J. Hoigné (1982). "Decomposition
of Ozone in water: Rate of Initiation by Hydroxide ion and Hydrogen Peroxide."
Environemental Science and Technology 16: 676-680.
Staehelin,
J. and J. Hoigné (1985). "Decomposition
of Ozone in Water in the Presence of Organic Solutes Acting as Promoters
and Inhibitors of Radical Chain Reactions." Environemental Science
and Technology 19: 1206-1213.
Tomiyasu,
H., H. Fukutomi, et al. (1985). "Kinetics
and Mechanism of Ozone decomposition in Basic Aqueous Solution." Inorganic
Chemistry 24: 2962-2966.
Trambarulo,
R., S. N. Ghosh, et al. (1953). Journal of Chemical Physics 21:
851-860.
Wadt,
W. R. and W. A. Goddard (1975). "The
Electronic Structure of the Criegee Intermediate. Ramifications for the
Mechanism of Ozonolysis." Journal of the American Chemical Society
97(11): 3004-3021.
Walling,
C. and G. El-Taliawi (1973). "Fenton's
Reagent. I. Reactions of Carbonyl Compounds and a, b-Unsaturated Acids."
Journal of the American Chemical Society 95(3): 844-847.
Weiss,
J. (1935). "Investigation on the radical HO2 in solution." Journal of
the Chemical Society Faraday Transaction 31: 668-681.
White,
H. M. and P. S. Bailey (1965). "Ozonation
of Aromatic Aldehydes." Journal of Organic Chemistry 30:
3037-3041.