HEMATOPATHOLOGY
Department of PathologyCornell University Medical College
INTRO TO HEMATOPOIETIC SYSTEM
HEMATOPOIETIC SYSTEM I
- Number of blood cells in the blood stream depends on three factors:
- There are two types of marrow:
Distribution of active marrow can be determined by administering radioactive iron:
Total marrow space in the adult is about 4 liters. About half of this is active. Total marrow space in the child is about 1.6 liters. It is nearly 100% active though.
Remember - The spleen and lymph nodes are also part of this system.
- Development of Marrow
The first hematopoietic stem cells appear in the yolk sac during the 3rd week of embryogenesis. At about the 3rd month of fetal life, some of the cells migrate to the liver which then takes over as the chief site of blood cell formation until just before birth. The spleen, lymph nodes, and the thymus also contribute. This is called extramedullary hematopoiesis. At about the 4th month of gestation the bone marrow spaces begin to become important as the source of cells. If a sufficient stress is placed on the adult, extramedullary hematopoiesis can be a compensatory mechanism.
HISTOLOGY
 3041 Table: Normal Hematopoeisis
3041 Table: Normal Hematopoeisis
Bone marrow is composed of jelly-like material dispersed among bony trabeculae and supported by a fine reticulin network. The marrow is perfused by a main nutrient artery with small terminal arterioles. The blood continues through venous sinusoids into a central sinusoid. The central sinusoid has a permeable basement membrane similar to the glomerular filtration barrier in the kidney. Red cells squeeze into the sinusoidal lumen, leaving their nuclei behind in the cellular matrix. Megakaryocytes line the sinusoids and discharge platelets individually or in ribbon-like streamers.
- Red cell line: Erythroblastic islands*** occupy the central marrow
spaces and surround a central histiocyte called a nurse cell. This cell
phagocytoses the nuclear material extruded by the maturing normoblast and
recycles iron. The nurse cell cytoplasmic processes will stain for iron.
 3004 Spleen, iron in macrophages,
Perl's stain. 3034 Iron stain.
3004 Spleen, iron in macrophages,
Perl's stain. 3034 Iron stain.
- Granulocytic line: Granulocytic/monocyte production initiates at the
osteoid-marrow junction with maturation/differentiation occurring
diffusely throughout the more central marrow spaces.
- Megakaryocytes: Mature megakaryocytes line the sinusoids and discharge platelets directly into the blood stream.
 3004
3004 3034
3034 If we consider the marrow to be the blood cell "factory," the "retail store" is the peripheral blood. The circulating cells usually reflect the marrow production. Outside-the-marrow events, however, such as extravascular hemolysis, may change the equation. (Peripheral blood smear morphology and interpretation will be dealt with in 17.2 - 17.4.)
- Cellularity
Examination of the marrow is usually done by needle biopsy of the iliac crest under local anesthesia. (The other possible biopsy site is the anterior superior iliac spine.) The procedure involves first aspirating some of the jelly-like marrow substance and smearing it onto a glass slide. Then a needle core biopsy is obtained. The information obtained from these is complementary. The aspirate smear is stained with Wright-Giemsa, iron and any other stains deemed necessary. Cellular composition and detail can usually be accurately assessed. The myeloid:erythroid (M:E) ratio -- something of a misnomer -- is obtained by counting a minimum of 200 cells at 100x magnification. The bone marrow biopsy is the more accurate for determining the cellularity and sometimes for processes involving the bone.
3024 Normal bone marrow,
undecalcified section, 25x, H&E.
The biopsy is also useful for evaluation of infiltrative processes.
1535 Non-caseating granulomas,
sarcoidosis of bone marrow, H&E. 15079 Bone marrow with granuloma,
Pott's disease. 1534 Metastatic breast carcinoma,
bone marrow, H&E. 3039 Metastatic carcinoma, breast 1ˇ,
100x, H&E.
The terminology employed, based on our knowledge of the pluripotential stem cell, divides the marrow cells into lymphoid and non-lymphoid lines. The term for all non-lymphoid cells is myeloid. This word includes the erythroid, granulocytic, monocytic/macrophage, and megakaryocytic lines. The normal values for a bone marrow aspirate (at NYH) are as follows:
The biopsy evaluates cellular production and is a function of age. A normocellular marrow for a child under 2 years is virtually 100% red marrow. A normocellular marrow for a mature adult is 40-50% fat, 60-40% hematopoietic elements.
1178 Normal bone marrow biopsy, low
power.
The healthy elderly population (<65 years) does not suffer a marked loss of
red marrow, contrary to common misconceptions.
Changes in the marrow may or may not be pathological. Some of the terms commonly used are:
- Hypercellular: An increase in one or more of the cell lines. The
mechanism is usually compensatory; for example, granulocytic hyperplasia
in response to infection, erythroid hyperplasia in response to anemia, or
megakaryocytic hyperplasia in response to hemorrhage. 1186 Erythroid hyperplasia, marrow
aspirate, Wright stain. 1222 Megakaryocytic hyperplasia, bone
marrow biopsy, H&E.
- Aplasia or hypoplasia: A diminution or complete loss of cellularity.
The cause may be idiopathic, iatrogenic (as with chemotherapy/radiation for
tumors), drugs, viral infections, etc.
- Lymphoid follicles: Although not present in younger adults and
children, their presence is normal in the elderly. They are not usually
present before age 50.
- Reticulin fibrosis:
Increased reticulin (Type III collagen). Determined with a silver stain. Only
determined on a biopsy. Graded as I through IV. The marrow may not be
aspirable. Some of the earlier stages may be reversible. 1218 Myelofibrosis, bone marrow
biopsy, reticulin stain.
- Myelofibrosis: Increased collagen, the type familar as a scar.
Determined with a trichrome stain (Mallory trichrome or Azan) on the
biopsy. This change is usually irreversible, as with any scar. Again, the
marrow cannot be aspirated.
- Osteosclerosis: Proliferation of the bony network. Again, this is irreversible.
- Hypercellular: An increase in one or more of the cell lines. The
mechanism is usually compensatory; for example, granulocytic hyperplasia
in response to infection, erythroid hyperplasia in response to anemia, or
megakaryocytic hyperplasia in response to hemorrhage.
 3024
3024 1535
1535 15079
15079 1534
1534 3039
3039 1178
1178 1186
1186 1222
1222 1218
1218
ERYTHROPOIESIS, HYPOPROLIFERATIVE ANEMIAS
Powers Peterson, M.D.
ERYTHROPOIESIS, HYPOPROLIFERATIVE ANEMIAS
FYI = For your Information
The quantity of medical information available is near infinite and it is not in the scope of this course to cover it all. Material following a FYI button serves to increase your understanding of the material or provide useful clinical correlations. However due to the already expansive amount of lecture information, you are not responsible for the material found in these FYI sections.
HEMOPOIESIS
The Pluripotential (Totipotential) Stem Cell
The pluripotential stem cell is defined as the precursor cell from which all erythrocytes, leukocytes, and megakaryocytes are derived (i.e. all blood cells have a common cell line of origin).

The common cell line theory is supported by the following evidence:
- Irradiated mice given infusions of cells bearing marker chromosomes
generate blood cells having the marker chromosome; i.e., the
derivative cells are clonal.
- The Philadelphia chromosome (Ph1)
Ninety per cent of patients with chronic myelogeous leukemia (CML) have a reciprocal translocation between chromosomes #22 and #9. The translocated portion of chromosome #22 is referred to as the Philadelphia chromosome, for the city in which its discoverers worked. In those cases lacking the cytogenetically obvious translocation, there is usually a rearrangemment of some portion of a region on chromosome 22 known as the breakpoint cluster region (bcr). Either the Ph1 chromosome or a bcr rearrangement is found in granulocytes, megakaryocytes, normoblasts and monocytes in CML.
- The proliferating cells in polycythemia vera (PV) as determined by
common G6PD-marker isoenzymes are clonal.
- Hemopoietic colonies in the spleen are established from single precursor cells.
Hemopoiesis: Quantitiative Data
In order to understand the mechanics of marrow production, we make the following assumptions:
- The hemopoietic sequence is considered as a series of compartments
and subcompartments representing definitive stages in the maturation
sequence.
- Reentry of cells into a compartment does not significantly affect the
population of that compartment.
- In some compartments (younger cells), there is active mitosis. In the
later post-mitotic compartments, mitosis is negligible.
- In mitotic compartments the generative cycle consists of four
successive stages: postmitotic rest, DNA synthesis, premitotic rest, and
mitosis.
- Total time required for progression through all four phases is the generation time. For a given cell line, the generation time is about equal in successive compartments.
The following is a schematic representation of the distribution and pools that exist along the maturation of the blood cells:
Erythropoesis
* maturation time in the bone marrow can be decreased to 2-3 days in response to an acute stress like an acute blood loss or hemolysis.
The bone marrow has three mechanisms at its disposal to respond to an acute decrease in circulating erythrocytes:
- The marrow can increase the number of cells in the erythroid pool by
increasing the number of stem cells that will differentiate into
erythrocytes;
- The marrow can decrease the time it takes for each erythroid cell to
reach maturation; or
- The marrow can release reticulocytes into the bloodstream earlier.
Granulopoesis

Stress on ANY cell line can cause the marrow to increase production of that line to 5-6 times normal in a short time; i.e. hours to a few days.
How is this production and distribution of blood cells normally reflected throughout the body--in the marrow, the circulating blood and the monocyte/macrophage (reticuloendothelial) system?
Erythropoiesis
Let's switch from talking about hemopoiesis in general to erythropoiesis specifically.
First, we need to define two terms.
Reticulocytes
These are young RBCs which have extruded their nucleus but still contain large amounts of RNA. (Reticulum is basically RNA.) At the reticulocyte stage, the cell stays one day in the marrow and another day in the peripheral circulation before losing its basophilia. The normal reticulocyte count is 1%. If higher, there's a stimulus to RBC production.
 3026 Reticulocytes, 100x, Wright stain.
3026 Reticulocytes, 100x, Wright stain.
- The average half-life is 4.8 hours (± 1.9 hours).
- The maturation time is prolonged in: thalassemia, pernicious anemia
(PA) in relapse, anemia of uremia.
- The count in the periphery is dependent on:
- Rate of release from marrow
- Degree of immaturity of newly released reticulocytes
- Rate of disappearance of reticulum.
- Rate of release from marrow
To visualize reticulocytes on a peripheral blood film a special (supravital) stain is necessary.
 3027 Reticulocytes, hemolytic anemia,
reticulin stain, 160x.
3027 Reticulocytes, hemolytic anemia,
reticulin stain, 160x.
INTRODUCTION TO ANEMIAS
Now that you have an understanding of erythropoiesis, recall that in the marrow there are three categories of cells:
- Stem cells: Both totipotential and unipotential (committed);
- Mitotic/proliferative and possibly storage cells; (there's virtually no
storage capacity for RBCs)
- Peripheral blood or circulating cells (non-nucleated red cells).
- RBCs are rounded to disc-shaped with an area of central pallor.
When examining a peripheral blood smear (PBS), find a normal lymphocyte
nucleus to compare with the RBCs. Both of them should be about 7µ in
diameter. Note the size and shape of the RBCs. 3023 Normal RBCs on PBS, Wright stain.
One evaluates the following:
- Cell size (microcytic, normocytic, or macrocytic)
- Cell-to-cell variability in both size and shape
(poikilocytosis, anisocytosis)
- Other abnormalities such as burr cells, target cells, inclusions (malaria, e.g.), nucleated RBCs, precipitates in reticulocytes, sickle cell, etc.
3033 RBC inclusion in malaria. 3032 Nucleated RBCs in severe
beta-thalassemia, Wright-Giemsa stain. 3036 Target cells -- iron deficiency,
Wright-Giemsa stain. 3029 Target cells and schistocytes --
Hb C disease.
- Cell size (microcytic, normocytic, or macrocytic)
- Hematocrit: An indirectly derived measurement on the Coulter counter.
The hemoglobin content is a directly derived measurement (accurately
determined on a spectrophotometer at 540 nm). The RBCs are described in
terms of the Hb content as hypochromic, normochromic or hyperchromic. In
terms of cell size, the RBCs are either microcytic, normocytic or
macrocytic.
- Anemia: Decreased oxygen carrying capacity of the blood. Anemia may
also be "defined" in terms of the Hb content (Hb < 12 g/dL in an adult male, <
11 g/dL in an adult female).
Anemias are classified into three pathophysiologic/etiologic categories:
- Blood loss (acute or chronic),
- Hypoproliferative (impaired production), or
- Hemolytic (increased destruction)
 3040 Table: Classification of Anemias
3040 Table: Classification of Anemias
- Blood loss (acute or chronic),
 3023
3023 3033
3033 3032
3032 3036
3036 3029
3029 3040
3040
ANEMIAS SECONDARY TO BLOOD LOSS
Blood Loss Anemias
- Acute: e.g., hemorrhage due to trauma, massive GI bleeding, or child
delivery. Usually the iron stores remain normal.
- Chronic: e.g., bleeding peptic ulcer or excessive menstrual bleeding. If there is not replacement of iron, the predictable result is depletion of iron stores and eventually a hypochromic microcytic anemia due to iron deficiency.
HYPOPROLIFERATIVE ANEMIAS
This is the largest of the three categories of anemias worldwide. To understand iron deficiency, one must first understand iron metabolism.
Iron Deficiency
- Normal Fe Metabolism
This is both common and correctable, so watch for it! Iron deficiency anemia affects approximately one third of the world's population. Iron regulation is very important. Too much can be toxic. A 70 kg man has a total of 4 g of iron in his body. About 2.5 g is in RBC's and about 1 g is in ferritin. It's also very important in non-hemopoietic tissues, where it's necessary for enzyme activity (e.g., the Krebs cycle).
Iron absorption is greatest in the duodenum and decreases progressively as one moves distally down the intestine. Iron is much more readily absorbed in its ferrous (Fe2+) form than in its ferric (Fe3+) form. Vitamin C can increase iron absorption by serving as a reducing agent to maintain iron as Fe2+.
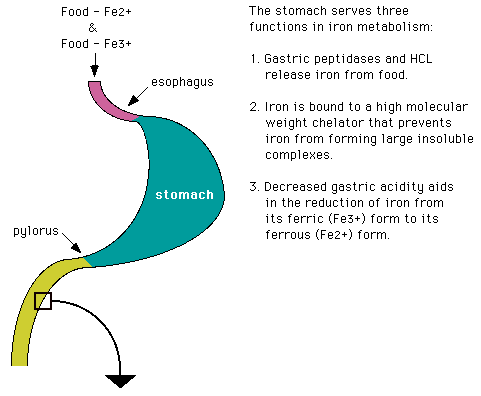
* Mucosal columnar cells have a 3-4 day lifespan before the cell gets sloughed. Any iron left in the cell at that time is lost to the body.
Iron requires transferrin, secreted by the mucosal cells of the duodenum, for absorption across the brush border. From there, plasma transferrin takes it to the tissue and blood cells. Tissue cells have membrane receptors that regulate how much transferrin, and therefore how much iron, they pick up. Lacking these receptors, the RBC maturing in the marrow just "gobbles up" the entire transferrin-iron complex.
The following is a diagram of the effect of low serum iron on iron absorption.

Humans are unusual in that we require heme iron as opposed to non-heme iron. We excrete about 1 mg/day of iron, which is about the same amount as we are capable of absorbing per day. In order to absorb that much, however, we must ingest 10-15 mg iron daily. Other animals can excrete 10 times as much iron, and they have no trouble absorbing enough to replace it. One more time: Our iron metabolism is a very tightly closed system.
- Reasons for iron deficiency:
- Chronic blood loss
- Inadequate intake
- Increased need: Growing children and pregnant women (children under 2 are almost always iron deficient)
- Chronic blood loss
- Factors that increase or decrease the absorption of iron
- Classification of Iron Deficiency Anemia
If iron intake is decreased, iron stores are first depleted without affecting the Hb content of the RBC's. This is a state of "iron-depleted erythropoiesis." When the RBC's start to lose iron, there is a state of "iron-deficient erythropoiesis."
RBCs first try to maintain the Hb content at the expense of the size of the cell. Mean corpuscular volume (MCV), an index of RBC size, begins to decrease (normal MCV = 80-98 fL). Mean corpuscular Hb (MCH), an index of the amount of Hb/RBC, remains unchanged (normal MCH = 24 - 36 pg).
Quantitative Iron Deficiency
- Low iron intake (nutritional)
- Diminished iron absorption
- Malabsorption syndromes
- Gastrectomy
- Pica
- Infection - Physiologically increased iron requirements
- Pregnancy and lactation
- Infancy - Excessive iron loss
- Chronic hemorrhage
- Hookworm infections
Inadequate Iron Utilization
- Chronic Disorders
- Infections (bacterial, mycotic)
- Collagen vascular disorders (RA, SLE, RF, etc.) Structure of heme
- Carcinoma (Ferroprotoporphyrin IX)
- Lymphoma
- Leukemia
- Thalassemia
(Note - In this iron overloaded state there may be "iron deficiency".)
When iron deficiency gets to the point where the RBC's can't maintain either the normal amount of iron or the normal size of the cell, then there are very small, very pale cells.
3035 Mycrocytosis due to iron
deficiency, 400x.A frequently seen RBC is the target cell, a small RBC with much less than normal Hb, resulting in a dark central area with a pale ring. Both the MCV and the MCH are low. If iron is not replaced and stores are markedly depleted, there is markedly ineffective erythropoiesis.
3036 Target cells.
- To diagnose iron deficiency:
- Take a thorough clinical history (menstruation, peptic ulcer, etc.).
- Examine the peripheral blood smear (PBS).
- Obtain appropriate laboratory studies:
- Laboratory Measurements of Iron Metabolism

*Variable.FYI-More on laboratory measurements
Serum Iron, TIBC, % Saturation
TIBC refers to serum tranferrin levels. % saturation refers to the percent of transferrin complexed with iron.
As iron stores approach depletion
When iron stores become depleted
Serum Ferritin:
Normally only minimal amounts are present in the bloodstream. Serum ferritin is measured by RIA. The range of normal is wide but rarely do serum ferritin levels fall below 10 µg/dL.
Serum ferritin levels are decreased during pregnancy and in iron deficiency anemia. In iron deficiency anemia levels are usually below 10 µg/dL with a median level of 4 µg/dL.
Free Erythrocyte Protoporphyrin (FEP):
Protoporphyrin is the immediate precursor to heme in heme synthesis (Protoporphyrin III + Fe2+ = Heme). Normal red blood cells have a small amount of protoporhyrin (15-80 ug of protoporphyrin per dL of red blood cells). In Iron deficiency anemia heme synthesis cannot progress past the protoporphyrin stage due to lack of iron to incorporate into the center of the complex. Protoporphyrin is therefore increased.
Early in iron deficiency FEP rises, often before the deficiency results in clinically overt anemia. FEP is also increased in the anemia of chronic disease, most sideroblastic anemias, and lead poisoning.
Lead poisoning blocks heme synthesis at 3 points:
- Lead inhibits the synthesis of d-aminolevulinic acid from glycine &
succinate.
- Lead also blocks the next step of 2 aminolevulinic acids to 1
porphobilinogen.
- Lead inhibits the incorporation of iron into the protoporphyrin ring to form heme.
Therefore in lead poisoning the bone marrow has abundant iron stores but cannot incorporate the iron into heme complexes. As a result FEP greatly increases.
In lead poisoning one sees porphyrin precursors in the urine, especially coproporhyrin III. The urine displays red fluorescence with Wood's glass after extraction with ether. The anemia of lead poisoning is usually normochromic but can occasionally be hypochromic. Peripheral blood smear is characterized by basophilic stippling and an increased retic count.
3002 Basophilic stippling.
FEP is decreased or normal in primary disorders of hemoglobin synthesis like thalessemia, in pyridoxine-responsive anemia, and in sideroblastic anemia where the defect is a block in heme synthesis prior to the synthesis of protoporphyrin.
TIssue Iron Stores:
Tissue iron stores are evaluated by bone marrow aspirate. The tissue specimen is then stained with Prussian blue which stains for hemosiderin. In Iron deficiency the bone marrow is depleted of its iron stores and therefore does not stain or stains very lightly. In the anemia of chronic disease, bone marrow iron stores are normal or increased.
3034 Iron stores.In sideroblastic anemia or thalassemia, bone marrow iron stores are significantly increased and the marrow stains intensely. This method of determination provides only a gross estimate of tissue iron stores but this is sufficient to differentiate iron deficiency from other hypoproliferative anemias.
- Lead inhibits the synthesis of d-aminolevulinic acid from glycine &
succinate.
- Low iron intake (nutritional)
 3035
3035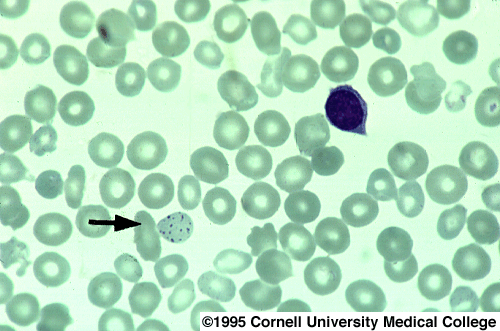 3002
3002MEGALOBLASTIC ANEMIAS: B12 AND FOLATE DEFICIENCY
- General Considerations
Essentially there is a dyssynchrony between nuclear and cytoplasmic growth. B12 and folate are required for DNA synthesis. A deficiency of B12 or folate produces a block in DNA synthesis, while RNA synthesis continues unhindered. Our B12 requirement is 1 µg/day, and the liver stores a 3-year supply. We get B12 from animal foods. Our folate requirement is about 50 µg/day, and we store a 3-4 month supply. This we get from leafy vegetables, bananas, cantaloupe, asparagus, chocolate, etc.
A megaloblast and macrocyte are not synonymous terms. Megaloblast refers to the cell's nuclear-cytoplasmic ratio. Macrocyte refers to cell size. While nearly all megaloblasts are macrocytes, there are multiple other causes of macrocytosis including {modified from Stein text book of medicine, pp.1037-8 (1987)}:
- Histology of Megaloblastic Anemia
Megaloblastic anemia looks "mega". Red cells are huge and oval (macro-ovalocytes), and there are hypersegmented PMNs (more than 5 lobes).
3037 Macro-ovalocytes, and target
cells.If the PBS has these huge cells and the MCV is over 110 fL, it's probably safe to assume the patient has a B12 or folate deficiency (or both). There are cases however, where the MCV is only slightly elevated but the patient is B12/folate deficient.
We also see reticulocytes in the peripheral blood, and a wide variation in the size and shape of the cells. The BM is undergoing erythroid hyperplasia, but it's ineffective because DNA synthesis is not proceeding normally. The myeloid:erythroid (M:E) ratio in the BM can get as high as 1:1. The chromatin of the erythroid cells is stranded rather than clumped, and cytoplasm is increased relative to the size of the nucleus.
3038 PA (E.dys synch).Because DNA is ubiquitous, all cell lines throughout the body are affected. However, the changes are most easily seen in the WBC and RBC lines.
- B12-Folate Metabolism


- B12/Folate Deficiency
B12 deficiency is caused by some of the same basic mechanisms that cause iron deficiency:
- Inadequate intake: Extreme poverty, very strict vegetarians,
alcoholics.
- Increased need: Pregnancy, parasites (the fish tapeworm D. latum),
hyperthyroidism, chronic blood loss.
- Impaired or inadequate absorption:
Folate deficiency is caused by:
- Inadequate intake (Chronic alcoholism).
- Increased need: Pregnancy, parasites, hyperthyroidism, chronic
blood loss.
- Inadequate absorption: Drugs.
- Hemodialysis. (Folate is dialyzable.)
Dilantin, barbiturates, and cholestyramine are examples of drugs that inhibit folate absorption. Also important to consider are the metabolic inhibitors: methotrexate, anti-malarials, oral contraceptives and trimethoprim.
Patients often do well with a low Hb level, as long as they get to that level slowly. The decreased oxygen-carrying capacity may result in:
- Fatigue
- Myocardial infarct (Hb Ĺ 5 gm/dL)
- "Megaloblastic madness" (patients actually present as psychiatric
cases)
- Subacute combined degeneration (only from B12 deficiency-- this is not a consequence of folate deficiency). Degeneration of the lateral spinothalamic tracts to the point that the patient can't walk. This is an irreversible change.
Classification of anemia caused by deficiency of vitamin B12 or folate
Vitamin B12 deficiency Folate deficiency - Caused by deficient intake (vegans)
- Resulting from lack of intrinsic factor
- Classic pernicious anemia
- After gastrectomy
- After destruction of gastric mucosa (acute leukemias, hemolytic anemias)
- Due to disease of small
intestine
- Blind loop syndrome
- Diseased or resected ileum
- In D. latum infection
3003 D. Latum eggs.
- Drug-induced malabsorption
- AIDS
- Dietary intake
- Increased folate requirements
- Pregnancy
- Infancy
- Increased cellular requirements
- Due to malabsorption of folate
- Drug-induced
- Steatorrheas
- Inadequate intake: Extreme poverty, very strict vegetarians,
alcoholics.
- Diagnosis of Megaloblastic Anemia
To make a diagnosis of megaloblastic anemia:
- Take a thorough clinical history.
- Examine the PBS for the appropriate changes in the RBCs and WBCs.
- The MCV should be very elevated.
- Measure red cell folate or serum B12 level.
- Perform a Schilling test, a nuclear medicine procedure that determines the
ability to absorb vitamin B12.
Schilling Test
The Schilling test is a 3 stage test.
Stage 1 - Give radioactive labeled vitamin B12 orally. Then screen the patient's urine for the radiolabeled B12.
Stage 2 - Give oral radiolabeled B12 with oral intrinsic factor and again screen the patient's urine for B12.
Stage 3 - Treat patient with 7-10 days of antibiotics. Then give radiolabeled B12.
- Take a thorough clinical history.
 3037
3037 3038
3038 3003
3003- Myelophthisic Processes
Myelophthisic: "Infiltration of the marrow," but the term does not identify the etiology of the process. Infiltration of the BM blocks the capacity of the hematopoietic cells to proliferate, usually on the basis of a space-occupying lesion. Examples include: neoplastic processes, granulomatous disease, myelofibrosis.
- Anemia of Chronic Disease
The anemia of chronic disease is a well known clinical entity, but the etiology is obscure. It is seen in hepatic, renal and endocrine disorders. A low serum iron is accompanied by a low or normal TIBC. There is a block in the reticuloendothelial utilization of iron and a shortened RBC survival time. The anemia is usually slight.
- Aplastic anemia
Aplastic anemia is the result of complete BM failure. The etiology is unknown in 50% of cases.
Aplastic anemia is a clinical disaster, and most patients will die from it. BM transplants have been successful in many more cases recently, but most patients "don't have the foresight to have an identical twin who doesn't have the disease."
Recombinant gene therapies (erythopoietin, granulocyte- and granulocyte-monocyte colony stimulating factor) may be helpful, but these are (for the most part) only in Phase 1 clinical trials (see diagram below) .
The defect is not known, but where it is not congenital, it is thought to be an acquired stem cell defect.

LEUKOPOIESIS, WBC KINETICS
Powers Peterson, M.D.
LEUKOPOIESIS AND NON-NEOPLASTIC WHITE BLOOD CELL DISORDERS
AIM
Today's discussion centers on three of the leukocytes present in peripheral blood and visualized on a PBS: granulocytes, monocytes and lymphocytes.
LEUKOPOIESIS
- Review: Totipotential and Multipotential Stem Cells (H/O,1/5/87)
- Committed stem cells: lymphoid line and myeloid line
- Lymphoid line: further differentiation into either
- Myeloid line: Further differentiations into:
Recall that the marrow preferentially supports granulopoiesis over erythropoiesis.
GRANULOCYTE AND LYMPHOCYTE KINETICS
- Granulocyte Kinetics
- Proliferative (mitotic) compartment: Those cells capable of
dividing (myeloblast, promyelocyte, myelocyte)
- Reserve (post-mitiotic) compartment : For the neutrophilic
series is largely composed of band neutrophils
- Circulating compartment: PMNs (a few band forms will be
present)
There are so-called "marginating PMNs" which are located literally along the sides of blood vessel walls. These PMNs may be considered as part of the circulating compartment. However, these PMNs are "released" into the circulation by a stimulus (trauma, steroids, etc.), so they act more as if they are part of the reserve compartment.
- Proliferative (mitotic) compartment: Those cells capable of
dividing (myeloblast, promyelocyte, myelocyte)
- Lymphocyte kinetics
- Same compartments as for the granulocytic series
- Although lymphocytes (L) circulate in the peripheral blood,
they also have a very important function in the thymus,
lymph nodes, spleen and sometimes in the liver.
- Outside of hematopoietic organs Ls are found at sites of chronic infection.
- Same compartments as for the granulocytic series
- Hormonal effects on the granulocytic/monocytic series
- Described in tissue culture but there is also in-vivo evidence.
- Androgens stimulate granulocyte production.
- No effect on granulocyte-monocyte series: adrenergic
antagonists, estrogen, GH, prolactin, progesterone, thyroxin.
- Dexamethasone, PGE-2 actively inhibit the growth and differentiation of the granulocyte-monocyte series.
- Described in tissue culture but there is also in-vivo evidence.
CHARACTERISTIC OF NORMAL GRANULOCYTIC CELLS
- Polymorphonuclear leukocytes (PMNS)
- Cell size = 10-15 µ ; multi-lobed nucleus (3-4 lobes)
separated by isthmus which should be less than one-half the
width of the lobe; if the isthmus diameter is greater and
there are only two lobes, the cell is called a band neutrophil.
- Many produced every day due to high utilization (1.6 x 109
cells/kg/day).
- Normally circulate 6-7 hours and the life span is about 56 hrs.
- Cytoplasmic granules contain acid phosphatase, acid
hydrolases, peroxidases, muramidase, lactoferrin and
collagenases; alkaline phosphatase is found in the
membrane portion
- EM: segmented nucleus; abundant glycogen; nuclear pores; golgi are sites of granule production
- Cell size = 10-15 µ ; multi-lobed nucleus (3-4 lobes)
separated by isthmus which should be less than one-half the
width of the lobe; if the isthmus diameter is greater and
there are only two lobes, the cell is called a band neutrophil.
- Eosinophils
- Small isthmus; bi-lobed nucleus sometimes obscured by
granules.
- Slightly larger than PMNs -- approximately 12-17 µ
- Important in allergy, atopy, drug and parasite reactions
(function not as well understood as that of PMNs)
- Granules contain peroxidases, acid phosphatase, arylsulfatase;
no alkaline phosphatase.
- EM: membrane-bound granules show a dense crystallized core.
- Most found in marrow & tissues; only 1% circulate.
- Mean T (1/2) = 8 hr in periphery.
- Small isthmus; bi-lobed nucleus sometimes obscured by
granules.
- Basophils
- Larger than PMN or eosinophil; heavily granulated.
- Larger, very basophilic granules contain histamine, chondroitin
sulfates, leukotriene B, eosinophil-chemotactic factors.
- Function: immediate hypersensitivity reactions -- binds IgE.
- Least common blood granulocyte.
- Larger than PMN or eosinophil; heavily granulated.
- Monocytes
- Diameter = 12-15 µ; (reniform) nucleus occupies half of the
cell area and is usually eccentrically placed.
- Many fewer cytoplasmic granules than PMNs; granules stain
both eosinophilic and basophilic and vary in size
- Cytoplasmic vacuolization.
- Irregular nucleus and chromatin irregularly distributed.
- Maturation takes 4-5 days; spends 1.5 days in circulation and
then resides in tissues (RE tissue in spleen, Kupffer cells in
liver, etc.) for up to 4 mo.
- EM: ruffling of plasma membrane.
- Maturation sequence: monoblast, promonocyte, monocyte, immature macrophage (nucleolus prominent), mature macrophage
- Diameter = 12-15 µ; (reniform) nucleus occupies half of the
cell area and is usually eccentrically placed.
CAUSES OF MONOCYTOSIS (NON-LEUKEMIC)
REACTIVE LYMPHOCYTIC DISORDERS
- Benign lymphocytic disorders: lymphocytosis
- Lymphocytes can have anomalies of both quality ( ex. neoplasms) and
quantity (usually too few)
- Pathological causes of Lymphocytosis: Please see page 5 of the transcript on The Spleen from 1/5/87 for a discussion of infectious mononucleosis (IM).
NON-NEOPLASTIC DISORDERS OF GRANULOCYTIC WBCS
Non-neoplastic white blood cell disorders can be generally classified as either qualitative or quantitative. We will first deal with qualitative defects.
Qualitative Disorders: Defective Chemotaxis or Abnormalities of Cell Response
Defective chemotaxis or random migration is associated with clinical syndromes generally characterized by deficient resistance to infection and specifically by deficient cellular defenses. These abnormalities are classified as abnormalities of chemotactic factors or abnormalities of cell response.
Qualitative Defects are rare but:
- Requirements for normal granulocyte release
- Examples of abnormalities of cell response
- Abnormalities of Chemotaxis
- Complement component deficiencies, esp. C3,
C5a and C567
- Kinin-generating system: Factors XII & XIII
Inhibition of complement activation: Patients with liver cirrhosis, autoimmune disorders (SLE)
Inhibition of chemotactic factors (not complement related): Prototype is Hodgkin's disease; Sezary syndrome, Wiskott-Aldrich syndrome
- Complement component deficiencies, esp. C3,
C5a and C567
QUANTITATIVE DISORDERS OF GRANULOCYTIC WBCS
Marrow responses to increased need
- Increased activity in stem cell compartment (circulating and in
marrow)
- Increased mitotic activity in proliferative compartment (bone marrow)
- Shortened generation time. In the RBC line, shortened
generation time results in increased numbers of peripheral
reticulocytes. In the WBC line, it results in increased
numbers of band neutrophils (to 10-15% of total
leukocytes); this change in the differential count is referred
to as a "shift to the left" meaning an increase in less
mature cells in the periphery.
- Accelerated bone marrow transit time
- Accelerated release from bone marrow
Note: When the bone marrow is stimulated because of a decrease in a blood cell, it usually responds by all five mechanisms at once.
Absolute vs. relative increases/decreases in the number of WBCs
- Relative increase in cells: The proportion of cells is increased
but the actual total number of cells is normal.
- Absolute increase: The total number of cells is increased; the percentage of the cell line may be normal or increased, depending on the total WBC count
Non-Neoplastic Causes of Granulocytosis or Neutrophilic Leukocytosis Note: With tissue necrosis (crush injury, burn) and in acute hemolysis and acute hemorrhage, the oxygen-carrying capacity of the blood is impaired and there is a loss of WBC's.
Non-Neoplastic Causes of Granulocytopenia or Neutropenia
- Infections can also decrease the number of WBCs. The
important ones: influenza, measles, hepatitis,
infectious mononucleosis, miliary TB, septicemia
- Hemopoietic disorders: pernicious anemia, aplastic anemia,
aleukemic leukemia, hypersplenism, Gaucher's disease
- Chemical agents: sulfonamides, antibiotics, antihistaminics,
analgesics, anticonvulsants (esp. dilantin), antithyroid
drugs, quinine, pronestyl, barbiturates, chlorpromazine,
colchicine
- Miscellaneous: ionizing radiation, SLE, cyclic neutropeni
Information on infectious mononucelosis and Gaucher's disease can be found in the transcript of the Spleen lecture of 1/5/87.
Diseases with a Normal Blood Count but with an Abnormal Blood Film
- These may be identified by observing a peripheral blood smear(PBS)
- Some examples (H-O,p.8)
- Hereditary Spherocytosis: A membrane disorder
- Hemoglobin C Disease: A large number of target cells observed
- Lead poisoning: Basophilic stippling of RBCs
- Parasite inclusions: Protozoal including Malaria
(Plasmodium sp.) and Babesiosis (Babesia sp.)
- Allergic reactions: Manifested by absolute eosinophil increase
- Hereditary Elliptocytosis: Another membrane disorder
- Others: Multiple Myeloma (unusual), Thalassemia trait,
consumptive coagulopathy (DIC), infectious mononucleosis.
- Agranulocytosis: An absolute decrease in granulocytes, sometimes seen in severe infections
- Hereditary Spherocytosis: A membrane disorder
- Pelger-Huet Disease
- Hereditary anomaly characterized by a bi-lobation of
normal neutrophils
- Hereditary form: No clinical consequences
- Acquired form: Very frequently heralds the development of a myelodysplastic acute leukemia
- Hereditary anomaly characterized by a bi-lobation of
normal neutrophils
- Disseminated Histoplasmosis: (Robbins,p.358)
- Very rarely, one can detect three small inclusions in circulating
PMNs
- This example points out the importance of examining a PBS
- Very rarely, one can detect three small inclusions in circulating
PMNs
©Copyright 1995, Cornell University Medical College
HTML by Adam Goldstein
Last modified July 26, 1995