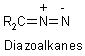STABLE CARBENE CHEMISTRY
Carbenes have played an important role in organic chemistry ever since the first firm evidence of their existence. They can be defined as divalent carbon intermediates, where the carbene carbon is linked to two adjacent groups by covalent bonds, and possesses two nonbonding electrons. These electrons may have anitparallel spins (singlet state) or parallel spins (triplet state).(1)
Fig. 1 – Representations of singlet and triplet carbenes
HISTORY OF CARBENES
The first attempts to prepare carbenes were made by Dumas and Regnault when they tried to prepare methylene by dehydrating methanol using phosphorus pentoxide or concentrated sulfuric acid.(1) Later, Butlerov obtained ethylene from the reaction of methyl iodide with copper and made the plausible suggestion that it arose by the dimerization of methylene.(1) Geuther made a proposal in 1862 that the basic hydrolysis of chloroform involved the intermediate formation of dichloromethylene, which has now been well established.(1)
A second period of carbene research was initiated by the discovery of isonitriles and fulminic acid derivatives. Nef, stimulated by his own work in this field proposed a "general methylene theory" which explained most substitution reactions by the sequence of a -elimination and addition.(1) Modern work in the field of methylenes began around 1910 with the investigations of Staudinger on the decomposition of diazo compounds and ketenes.(2) However, it was not until 1950 that the recent growth in divalent carbon intermediates began.
The organometallic chemistry of carbenes was initiated with the synthesis of stable carbene complexes by Fischer and Maasbol in 1964(3). It was not until 1968 that the first homonuclear carbene complex was reported(4), and isolation of a carbene containing the parent methylenegroup was not achieved until 1975(5). The high stability of diaminocarbenes was demonstrated in 1991 when Arduengo described the synthesis and structure of 1,3-diadamantylimidazol-2-ylidene, the first unambiguously stable carbene.(4)
GENERATION OF CARBENES
There are numerous ways of generating carbene intermediates. Some of the most general routes are summarized in the table below.
|
Precursor |
Condition |
Products |
|
|
Photolysis, thermolysis, or metal-ion catalysis |
R2C: + N2 |
|
|
Photolysis or thermolysis, diazoalkanes are intermediates |
R2C: + N2 + ArSO2- |
|
|
Photolysis |
R2C: + N2 |
|
|
Photolysis |
R2C: + R2C=O |
|
R2CH-X Halides |
Strong base or organometallic compound |
R2C: + BH + X- |
|
a -Halomercury compounds |
Thermolysis |
R2C: + R’HgX |
Table 1 – General Methods for the synthesis of carbenes(7)
TRIPLET CARBENES
Electrophilic carbenes :CR1R2 with substituents R1 and R2 that are not electron donors were originally regarded as being too reactive to be isolated. As these triplet carbenes cannot be stabilized through thermodynamic effects, they have to be rendered unreactive by steric protection.(13) Considerable progress was made by Tomioka et al., who stabilized diphenylcarbenes with methyl or chloro substituents in ortho positions.(13) In 1995 they synthesized 2,2',4,4',6,6'-hexabromodiphenylcarbene by photolysis of the parent diazo compound. (fig. 2) In glassy and completely fluid 2-methyltetrahydrofuran, the compound is stable without significant decomposition.(13) Triplet carbenes exhibit radical-like reactivity
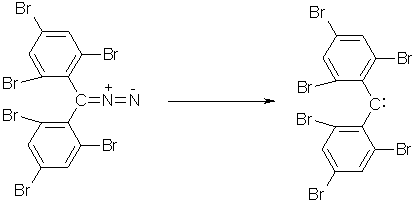
Fig. 2 – Synthesis of 2,2',4,4',6,6'-hexabromodiphenylcarbene by photolysis of the parent diazo compound
SINGLET CARBENES
Heteroatom donor groups on a carbene center render the originally degenerate orbitals on carbon unequal in energy, thus enhancing the nucleophilicity of the carbon atom and the thermodynamic stability. The singlet-triplet splitting correlates with increasing electronegativity of the p -donor substituents X and Y in carbenes of the type :CXY. Although several combinations of heteroatoms (O, S, N) are conceivable, only singlet carbenes with two nitrogen atoms (amino groups) were isolated so far ("carbon diamides"). These singlet carbenes have a pronounced low energy HOMO and a high energy LUMO. Because of the lower electronegativity of carbon, they are stronger electron-pair donors than amines., with the electron-accepting capability even more significant than that of boranes. Since the amino groups are p -donating (mesomeric) and s -withdrawing (inductive), 2,3-dihydro-1H-imidazol-2-ylidenes benefit from a "push-pull" effect. Whereas triplet carbenes exhibit radical-like reactivity, singlet carbenes are expected to show nucleophilic and electrophilic reactivity due to the s -type lone pair and the vacant p orbital.
STRUCTURE AND REACTIVITY OF CARENES
Depending on the mode of generation, a carbene may be initially formed in either the singlet or triplet state, no matter which is lower in energy.(7) Because of their paramagnetic character, triplet carbenes can be observed by electron spin resonance spectroscopy, provided they have sufficient lifetimes.(7) Molecular orbital calculations lead to the prediction of H-C-H angles for methylene of 135o for the triplet and 105o for the singlet.(7) Experimental determinations of the geometry of CH2 tend to confirm the theorectical results. The H-C-H angle of the triplet state, as determined from the EPR speactrum is 125-140o (7). The H-C-H angle of the singlet state is found to be 102o by electronic spectroscopy.(7)
The importance and extent of aromatic-delocalization for the carbene, has remained controversial.(3) Although the original synthesis of the carbene was rationalized by assuming that delocalization of the nitrogen lone pairs into the empty p orbitals would stabilize the electron-deficient carbene center, the majority of early computational work concluded that this type of delocalization was negligible or, at best, only a minor stabilizing factor.(3) However, failure of these early calculations to identify significant bonding between the C=C of the unsaturated ring carbons caused these publications to come under heavy scrutiny.(3) Spectral and computational studies conducted by Lehmann et al. indicates a high degree of delocalization in the heterocyclic ring of the following compound(3)

Further, the most significant mode of stabilization of the divalent site is the strong N – C – N delocalization.(3)
The following section details the synthesis and structure of various stable carbenes.
1,3-di-1-adamantylimidazol-2-ylidene (Arduengo’s carbene)
Carbene 1, 1,3-di-1-adamantylimidazol-2-ylidene was formed from the deprotonation of 1,3-di-1-adamantylimidazolium chloride (2) in THF at room temperature with catalytical dimsyl anion (-CH2S(O)CH3) in the presence of 1 equivalent of sodium hydride.(6) The deprotonation could have also been accomplished with potassium tert-butoxide in THF to give a 96% yield of 1.(6)
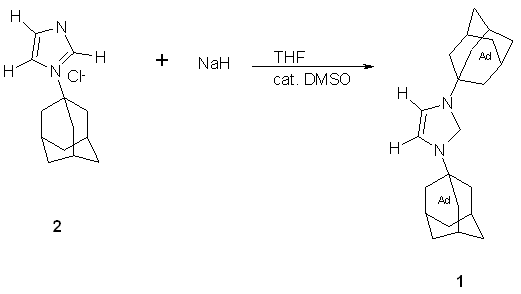
Fig. 3 – Formation of 1,3-di-1-adamantylimidazol-2-ylidene(6)
|
|
Bond length |
|
Bond angle |
|
C2 – N1 |
136.7 |
N1 – C – N3 |
102.2 |
|
C2 – N3 |
137.3 |
C5 – N1 – C2 |
112.1 |
|
C4 – C5 |
133.8 |
C4 – N3 – C2 |
112.3 |
|
N1 – C5 |
138.2 |
N1 – C5 – C4 |
107.2 |
|
N3 – C4 |
138.6 |
N3 – C4 – C5 |
106.2 |
|
N1 – C1-Ad |
148.2 |
C2 – N1 – C1-Ad |
123.4 |
|
N3 – C5-Ad |
148.5 |
C2 – N3 – C1-Ad |
122.1 |
Table 2 – Selected bond lengths (pm) and Angles (deg) in compound 1(6)
Several features were revealed in the X-ray structure of the above carbene. One was the small N1 – C2 – N3 angle at the carbene center, which was significantly reduced from the typical range of values (108.5-109.7o) for the corresponding angle in imidazolium salts. Further, the length of the C2 – N1 bonds were significantly increased from the value of 132 pm found in imidazolium salts. The largest structural changes were localized at the carbene center, which suggested a diminished p -delocalization in 1 compared to imidazolium salts.(6)
There are several reasons for the stability of carbene 1. First, the large electronegativity of nitrogen stabilizes the lone pair on the carbon center in the plane of the ring through an inductive s -effect.(3) Second, the unoccupied carbon(II) orbital can be stabilized by mesomeric interaction with the nitrogen lone pairs, which provides about 70 kcal/mol of stabilization.(3) A further 26 kcal/mol of stabilization is predicted to be associated with an overall 4n + 2 aromatic Hückel configuration in the unsaturated ring.(3) The planar geometry and the proton NMR spectrum, which exhibits strongly deshielded protons at 6.91 ppm, both strongly support the proposed aromatic nature of the heterocycle. Bulky substituents bound to the nitrogen centers have been shown conclusively to play a vital role in the stability of isolatable carbenes(8).
Bis(diisopropylamino) carbene
The stability of Arduengo’s carbene was attributed, along with other reasons, to its aromatic stabilization. However, the isolation of bis(diisopropylamino)carbene by Alder et. Al demonstrated for the first time that neither aromatic stabilization nor the constraints resulting from ring geometry were necessary to obtain stable diaminocarbenes.(12)
The isolation of bis(diisopropylamino)carbene (N,N,N', N'-tetraisopropylformamidinylidene), 3, as a stable solid was accomplished by the deprotonation of N,N,N',N'-tetraisopropylformamidinium chloride, 4, with lithium diisopropylamide in THF.(12) This reaction led to the formation of 3 in 55% yield. Diaminocarbene 3 sublimed readily and was stable under rigorously dry nitrogen, but appeared to be significantly more sensitive to oxygen and moisture than other species of the same type handled.

Fig. 4 – Formation of bis(diisopropylamino)carbene (N,N,N', N'-tetraisopropylformamidinylidene)
The X-ray crystal structure of the molecule showed that it had approximately C2 symmetry; the N – C – N angle (121.0o) was much larger than in any previously studied diaminocarbene derivative.(12) The nitrogen atoms were planar, but the C – N – C angles were strongly distorted by the repulsive interactions between the isopropyl groups.(12) The barrier to N – C rotation gave proof of substantial double bond character. The 13C NMR chemical shift of the carbene center at d =255.5 was 30-40 ppm downfield of the carbene carbon chemical shifts for imidazol-2-ylidenes, and may have been related to the increased N – C – N angle.(12)
In solutions containing both 3 and 4, separate NMR signals were seen for the two species, although there was some indication of broadening at higher temperatures.(12) This indicated that proton transfer between 3 and 4 was slow on the NMR time scale, due to the the steric hinderance of compound 3.(12) In summary, compound 3 was the first acyclic diaminocarbene, with its C – N bonds having substantial double bond character.
[Bis(diisopropylamino)phosphino[trimethylsilylcarbene] (Bertrand’s carbene)
The starting point in this development was the observation made by T. Curtius that a -bis(diazo) compounds spontaneously lose N2 to give acetylenes.(9)
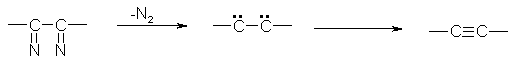
Fig. 5 – The loss of nitrogen in a -bis(diazo) compounds to give acetylenes
Bertrand was able to show that the thermolysis of [bis9diisopropylamino)phosphino](trimethysilyl)diazomethane (5) leads, after loss of nitrogen, to a stable compound 6 (1,1-Bis(diisopropylamino)-2-trimethylsilyl-1-phosphaacetylene).(10) While the NMR data and reactivity of 6 towards trimethylsilyl chloride, dimethyl sulfoxide and trimethylsilyl azide were characteristic for a phosphorus-carbon multiple bond, the carbene character of 6 was identified based on cycloproponation reactions, oxirane formation, and [1+1] addition to isocyanides.(11)
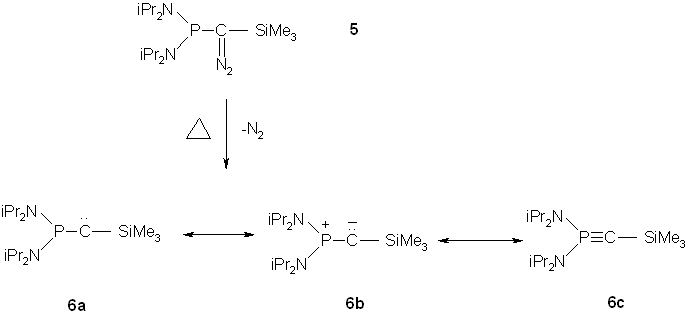
Fig. 6 – Formation of (1,1-Bis(diisopropylamino)-2-trimethylsilyl-1-phosphaacetylene)
The initial studies of 6’s reactivity showed that it could undergo the following reactions:

Fig 7 – Reactions carried out by (1,1-Bis(diisopropylamino)-2-trimethylsilyl-1-phosphaacetylene)(9)
Reactivity typical for carbenes involved the intramolecular C-H insertion of 10 at 3000 to form the tetrahydro-1,2-diazophosphole 9.(9) Further, the stereospecific [2+1] cycloaddition of 6 to electron-poor olefins such as dimethyl fumarate (6 à 10) demonstrated the compound’s character as a nucleophilic carbene.(9) This character also accounted for 6’s silylation to give the methylenephosphonium fluoromethanesulfonate 11.(9) A preliminary conclusion, based only on the reactivity of 6, was that the nonbonding electrons on phosphorus were involved in stabilizing 6 as shown by formula 6b.(9) The silyl group also acts to stabilize the negatively charged center. Moreover, steric shielding probably played a role as well.(9)
USES OF CARBENES
Stable carbenes, silylenes, and germylenes are valuable new building blocks for the synthetic chemistry of the respective elements. Further, the high volatility of these compounds allows their use as CVD precursors. Following is a short list of the many reactions that carbenes play an active role in.
It has been shown by Bertrand et. al that the stable phosphanyl(silyl)carbene 12 undergoes formal 1,2-addition reaction with protic reagents as well as with Lewis acids, giving rise to the corresponding phosphorus ylides. Further, the reaction of 12 with alkylythium reagents leads to the lithium phosphonium ylide 13.(12)
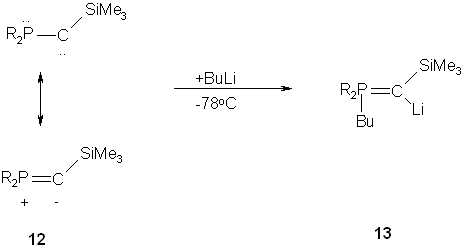
Fig. 8 – Formation of phosphorus ylides through reaction with carbenes
Compound 13 is highly moisture sensitive and is easily transformed into the corresponding phosphonium ylide 14. It also reacts at low temperature with electrophiles such as methyl iodide and chlorodiphenylphosphane to give the corresponding ylides 15 and 16 respectively.
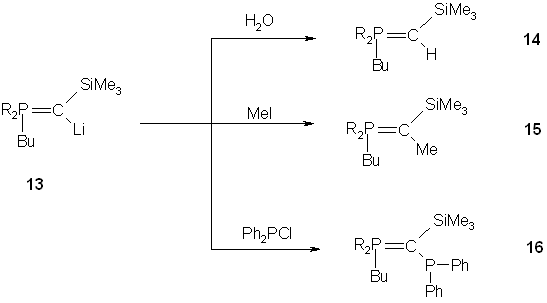
Fig. 9 – Reactions of phosphonium ylides synthesized via carbene intermediates
Electrophilic carbenes are also known to react with carbonyl groups through the oxygen lone pair to give carbonyl ylides (R2C=O – CX2), which are usually characterized by [3+2] cycloaddition, but can even be isolated.(11) Compound 12 does not react with dimethyl ketone but readily adds to benzaldehyde and cinnamaldehyde, producing oxiranes 17 and 18.(11) After treatment with elemental sulfur, 17' and 18' were isolated in 80% and 82% yields respectively, as only one diastereoisomer.

Fig. 10 – Formation of oxiranes via phosphanyl(silyl)carbene
Carbene 12 also reacts with t-butyl isocyanide to give the corresponding ketene imine 19, which is isolated after sulfuration, as 19' in 90% yield.(11) The reaction of 12 to give 19 can be considered as a carbene-carbene coupling reaction.

Fig. 11 – Formation of ketene imines via phosphanyl(silyl)carbene
N-Heterocyclic carbenes acting as nucleophiles have manifold catalytic application in synthetic chemistry as well as in nature: Breslow showed in the context of ylidene-catalyzed benzoin condensations that the vitamin B1 enzyme cofactor thiamin (20), a naturally occuring thiazolium salt, plays a key role in biochemistry.(14) In basic aqueous buffers the active catalyst of this reaction is a 2,3-dihydrothiazol-2-ylidene, 21. (fig. 12)

Fig. 12 – Structures of B1 enzyme cofactor thiamin (20) and active catalyst of this reaction is a 2,3-dihydrothiazol-2-ylidene (21).
Carbenes are also used for the catalysis of a wide variety of reactions, such as the hydrosilation of alkenes and alkynes.(14) For example, the hydrosilation of terminal alkenes catalyzed by rhodium-carbene complexes such as [RhCl(h 4-1,5-cod)L], [RhCl(PPh3)2L], and [RhCl(CO)(PPh3)L] leads to selective antimarkovnikov addition of the silane.

Fig. 13 – The hydrosilation of a terminal alkene catalyzed by a rhodium-carbene complex
The hydrosilaition products are obtained in yields of up to 98%, depending on the silane and the rhodium-carbene catalyst.
Carbenes are also used to catalyze the hydrosilation of ketones, using rhodium carbene complexes, where the mixed phosphane carbene derivative [RhCl(PPh3)2L] shows the best performance.(14) The hydrogenation of olefins is carried out using mixed carbene-phosphane complexes, for example [RhCl(PPh3)2L] and [Ru(Cl)L(PPh3)2].(14)
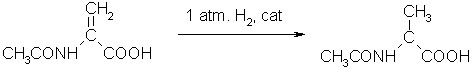
Fig. 14 – The hydrogenation of olefins using mixed carbene-phosphane complexes
Palladium-carbene complexes are used as catalysts for the Heck olefination of aryl halides, according to the reaction below(14)

Fig. 15 – The use of palladium-carbene complexes for catalyzing the Heck olefination of aryl halides
Carbenes are also used as catalysts in the hydroformylation of olefins, and in furan synthesis. The furan synthesis involves a ruthenium(II) carbene complex of the general type [Ru(p-cymene)Cl2L]. For example, in a catalytic reaction (Z)-3-methylpent-2-en-4-yn-1-ol is converted into 2,3-dimethylfuran.(14)
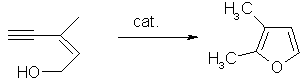
Fig. 16 – Furan synthesis using a ruthenium(II) carbene complex
CONCLUSION
In conclusion, there are three main reasons why carbenes have become the focus of active research:
Based upon these facts, and their renaissance in the past several years, it can be said with certainty that carbenes will play a major role in tomorrow’s organometallic chemistry.
REFERENCES