Hemophilia | The
History of Hemophilia | Types of Hemophilia |
Other Disorders
Severity | Inheritance
| Treatment | Prophylaxis
| Inhibitors | Physiotherapy
| Cure
Hemophilia
Hemophilia is the oldest known hereditary
incurable bleeding disorder. In a normal person the blood clots in few
minutes from the suffered wound and the healing starts but in the case
of Hemophilia patients, one of the clotting factor needed for the blood
clotting is defficient or absent which hampers the clotting resulting in
prolonged bleeding from minor cuts & bruises than a normal person.
The failure of the body to produce the required clotting factors is Hemophilia.
It affects mostly males whereas females act as carriers but there are some
rare cases in the world where females are found to be suffering from this
genetic disease. Naturally, women hemophiliacs are rare because it takes
two defective X chromosomes in order for the condition to be seen. Approximately
1 in 10,000 males have Hemophilia. A person with Hemophilia suffers from
internal bleeds in the joints and muscles which cause severe pain, swelling
which if untreated lead to progressive crippling. The major cause of disability
in hemophilia patients is chronic joint disease - "arthropathy" caused
by uncontrolled bleeding into the joints. Life-threatening hemorrhage is
a constant risk.
History of Hemophilia
The history of Hemophilia is probably
as old as the history of man, but the disorder would not have been so widely
recognized if it had not been for the descendants of Queen Victoria of
England. Victoria was the granddaughter of George III. She was born in
1819, the only child of Edward, Duke of Kent, and Victoria, Princess of
SaxeCoburg. She succeeded her uncle, William IV, to the throne in 1837
and three years later married Albert, son of the Duke of Saxe-Coburg-Gotha.
They had nine children, 4 sons and five daughters. Of the nine, one son,
Leopold, had hemophilia, and at least two daughters, alice and Beatrice
were carriers.
Leopold was the youngest son. During his
birth on 7th April 1853, chloroform was administered to Victoria by Dr.
John Snow of Edinburgh, a landmark in the history of early general anesthesia.
Leopold was clinically severely affected. He bruised easily and suffered
recurrent haemostasis which left him with a permanantly affected knee.
When he was 15 the Queen bestowed on him with the Order of the Garter,
wishing to give him his encouragement and pleasure, as he has so many
privations and disappointment. At 26 Leopold was prevented from representing
the Queen at the opening of the first Australian International Exposition.
Leopolds place in Australia was taken by brother, Alfred, who was shot.
The bullet was removed and he recovered, the probe used in the operation
still hangs on the wall in the hospital named after him in Sydney. If it
had been Leopold there is little doubt that he would have bled to death,
the victim of assasination.
Leopold married in 1882 at the age of 29
years. His bride, Helena of Wedbeck, bore him two children, a daughter,
Alice and a son. Before the son was born Leopold died after fall in Cannes;
he was 31. His daughter Alice was, of course, a carrier and at least one
of her descedants, Rupert, was haemophilic. Rupert died after a car crash
in 1928.
Queen Victorias carrier daughter were
Alice, born in 1843, and Beatrice born in 1857. Princess Alice had seven
children, of whom one Federick had hemophilia, and two, Alix and Irene
were carriers. Frederick, or Frittie, died when he was 3 year old after
a fall from a window. Alix became Czarina of Russia when she married Nicholas
in 1894, taking the name Alexandra Fedorovna. One child, Alexei, had hemophilia.
But we shall probably never know if any of his sisters were carriers. When
the remains of the family were found and identified by genetic fingerprinting
after the disintegration of the Soviet Union two bodies, including that
of Alexis, were missing.
Princess Irene married her cousin Henry
of Prussia and of their three sons, two were affected. Waldemer died in
1945 aged 56. His brother Henry died of bleeding aged 4, after a short
life hidden from society.
The second of Victorias carrier daughters,
Beatrice, passed the hemophilia gene the Spanish Royal family following
her marriage to the Prince of Battenberg in 1885. The family name was changed
to Mountbatten during the First World War at the instigation of George
V. The Mountbattens had four children. Leopold and Maurice had Hemophilia.
Incredibly both served in the war, Maurice being killed in action at Ypres
in 1914. Leopold lived to 33, when he died after an operation.
Their sister, Vicotria, married Alfonso
XIII of Spain, bearing him five sons and two daughters. The youngest son,
Gonzalo, had hemophilia, he died after a crash in 1934. His hemophilic
elder brother, Alfonso, also died after a crash in 1938.
With the death of Waldemar in 1945, the
hemophilia gene which plagued the family life of royalty in three countries
for almost 100 years and changed the course of European history, appears
to have become extinct. There remains a possibility that the disorder could
reappear in the descendants of one of the Queen Victorias daughters or
of her only affected son, Prince Leopold. Edward VII did not have haemophilia,
therefore the present British Royal family canot have inherited the disorder.
Hemophilia spread through the Royal Houses of Europe as monarchs arranged
marriages to consolidate political alliances. We can trace the appearance
of hemophilia as it popped up in Spain, Russia, and Prussia. Hemophilia
is also known as the Royal Disease because of its deep connections with
the Royal families around the world.
Types of Hemophilia
There are many blood clotting ingredients
which are called as factors. Factors is nothing but proteins manufactured
in the body and most of them are made in the liver. The most common types
of Hemophilia are Factor VIII and Factor IX which is also referred as Hemophilia
A and Hemophilia B respectively. Hemophilia A is 5 times more common than
Hemophilia B. All these factors are identified with Roman numbers. The12
main clotting factors with their alternative names are :
|
I
|
fibrinogen |
VIII
|
AHF - anti-hemophilic
factor AHG - anti-hemophilic globulin |
|
II
|
prothrombin |
IX
|
Christmas factor |
|
III
|
tissue extract |
X
|
plasma thromboplastin
antecedent (PTA) |
|
IV
|
calcium |
XI
|
Hageman factor |
|
V
|
|
XII
|
|
|
VII
|
|
XIII
|
fibrin stablizing
factor |
Other Clotting Disorders
Factor I
(fibrinogen) deficiency : Joint
bleeds are uncommon, but there is more likelihood of oozing from small
wounds than in hemophilia. Easy bruising will occur.
Factor II
(prothrobin) deficiency : Nosebleeds,
heavy periods, and easy bruising are features. Joint bleeds are rare.
Factor V
deficiency : Easy bruiding, nose
bleeds, heavy periods and joint and muscle bleeds are experienced by severely
affected people.
Factor VII
deficiency : Effects are usually
mild but include nosebleeds, heavy periods and bruising. Joint bleeds are
rare.
Factor X
deficiency : Bruising, joint bleeds,
and heavy periods occur in people with this deficiency.
Factor XI
deficiency : Affected people often
have Jewish ancestry. Nosebleeds and heavy periods are experienced.
Factor XII
deficiency : This deficiency is
usually diagnosed only when blood taken for some reason fails to clot in
normal time. Surprisingly, people with even a severe deficiency have little
or no trouble from bleeding.
Factor XIII
deficiency : Clots form in the
normal time but tend to break down later as they are not stabilized. This
leads to a delay in the healing after injury. Females with severe deficiency
have frequent miscarriages. Factor deficiency may present early in life
with oozing from the umbilical cord. People with severe factor deficiency
should be treated with monthly prophylaxisn with factor concentrate to
prevent any bleeds altogether.
Von Willebrand Disease : Von Willebrand's
Disease is a disease in which there is a defficiency of both factor VIII
& in platelet function with consequent prolongation of the clotting
and bleeding times is very common. Unlike Hemophilia both Males & Females
are affected because of this disease but it is usually much milder than
Hemophilia. People with vWD bleed for longer than normal. People
may bleed from small cuts, nose bleeds, bleed from the bowels and gums.
Women can experience heavy periods. Lack of vWF thus leads to condition
like hemophilia. The treatment comprises of decompressin (DDAVP) or a Factor
VIII concentrate with von Willebrand factor. Cyclokapron is often helpful
in stopping the breakdown of a blood clot.
Severity
The severity of hemophilia is determined
according to the amount of the clotting factor in the blood. Most of the
hemophilia patients have less than one percent of the normal levels of
factor in the blood which puts them in the category of severe hemophilics.
Patients upto 5% factor levels are regarded as moderate hemophilics. Above
5% is mild hemophilia.
Inheritance
Hemophilia is an inherited disorder which
means it can be passed on to the next generation of the family through
genes. Humans have 46 chromosomes out of which 2 determine the sex of an
individual. They are called as X and Y. Inheritance of 2 X chromosomes
denotes Female (XX) and 1 X chromosome and one Y chromosome denotes Male
(XY). The genes of hemophilia are on X chromosome. The following figures
expalin inheritance possibilities.
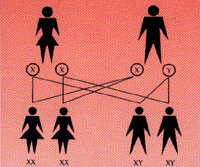
|
Human body is made of many cells. These
contain the genes inherited from the parents which control the functioning
of the body. Two of which determine sex. Females (XX) have 2 "X" chromosomes,
whereas Males (XY) have one "X" & one "Y" chromosome. One chromosome
from each parent determines child's sex. There are 4 possible combinations
:- |
 |
When the mother is a carrier and father
is unaffected, there is a 50/50 chance for their children i.e. their son
having Hemophilia and their daughter becoming carriers. |
 |
When the father has hemophilia and mother
is not a carrier, all their daughters will be carriers. Sons and their
future generations would be normal. |
Treatment
The only temporary treatment available
today is the expensive clotting factor concentrates which help in controlling
the bleed. It is also known from international studies that crippling joint
involvement can be avoided through periodic and regular transfusion. The
absent or deficient factor is infused through intravenous injections into
the patients body which helps in controlling the bleeding for few hours.
The amount of units to be infused is determined as per the severity of
hemophilia and the weight of the patient. These factors are produced from
blood plasma of human donors and some cases from pigs. Some are even made
synthetically by bio-engineering. All these products undergo stringent
testing and measures so that the end user gets a viral free safe product.
The availibility of these factor injections have revolutionised the Hemophilia
treatment world over. It is definitely expensive but the risks of viral
infections is very low unlike the fresh frozen plasma & cryoprecipitates.
Prophylaxis
Traditional treatment of hemophilia has
involved "on-demand" treatment, meaning that patients are treated with
factor replacement only after bleeding symptoms are recognized. These bleeds
ultimately result in severely impaired joints. Several European countries
are treating hemophiliacs by periodic infusion (prophylaxis) regardless
of bleeding status. This approach helps in maintaining the factor level
high enough that bleeding, joint destruction, and life-threatening hemorrhage
are almost entirely avoided. There are, nevertheless, serious disadvantages
such as the need for frequent infusions, the requirement for almost continuous
access to veins by catheters, and the high cost of factor.
Inhibitors
While current treatment has greatly improved
the outlook for most hemophiliacs, the development of antibodies (inhibitors)
that block the activity of the clotting factors has complicated treatment
for some patients. When inhibitors are present in large amounts, the patient
may require very high and expensive quantities of transfused clotting factors
to stem bleeding, and, in some instances, even that may not be effective.
The factor products produced through biotechnology have been found to cause
inhibitors in only about 5 percent of patients and are, thus, safer in
this respect. Nevertheless, these inhibiting antibodies will remain a concern
for hemophilia patients.
Physiotherapy
Physiotherapy plays an important part
in the lives of people with Hemophilia. It not only strengthens the muscles
and joints but also keeps you fit both mentally and physically. Strong
muscles and joints decrease the chances of bleeds considerably. Regular
exercises should be undertaken after taking proper advice from your local
Physiotherapist who is aware of Hemophilia.
Cure
Lots of research is going on throughout
the world, but a permenant cure seems to be a distant possibility. Although
treatment for hemophilia has become safer, therapeutic products are still
not risk free. Hemophilia is known to be caused by defects in the genes.
The challenge is to transfer normal genes into a patient so that they will
produce the normal clotting protein. A small amount of active factor produced
by the patients own body will correct the disease. Although much remains
to be studied before such treatment can be offered to patients, there have
been a number of studies done in animals such as mice and dogs in which
a factor gene has been inserted and has produced the proper blood product
for periods that exceed one year. Major issues that remain to be resolved
include the low level of production of the clotting factor, reduction of
immune reactions that stop the production after a period, and development
of ways to insert the gene directly into the body without manipulating
cells outside the body. Until recently, dogs with naturally occurring hemophilia
were used for testing of gene therapy techniques; however, the number of
such animals is very limited. At the present time, there are sufficient
indications that gene therapy will ultimately be this cure. Hemophilia
will likely be among the first genetic diseases to be successfully treated.
There have been reports of success in gene therapy which may cure Hemophilia
B. Let us all hope and pray that this "incurable disease" becomes "curable"
soon.