1 THE INSTITUTE
2 THE PROJECT
3 theoretical background
3.1 An introduction to glycobiology (1)
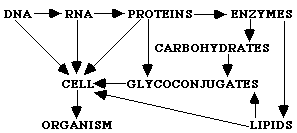
The essentials of contemporary molecular biology are based on the expression of biological information through translation of the DNA configuration into proteins, which form the cells that construct an organism. This theorem can be displayed as follows:
DNA
Æ RNA Æ PROTEIN Æ CELL Æ ORGANISMAlthough this theorem is correct and very often used, it is not complete. Two more major classes of molecules, lacking in this theorem, are indispensable to create a cell: lipids and carbohydrates. Especially carbohydrates are essential when it comes to creating multicellular organs and organisms, because they play an important role in cell-cell interactions and interactions with the ECM. Carbohydrates also mediate interactions between organisms (e.g. between host cells and bacteria, parasites or viruses (2)). The main theorem can thus be extended as follows:
Regardless the importance of several polysaccharides in the clockwork of an organism, the studies of glycans lingered for a long time compared with the other major classes of biomolecules during the first part of the modern revolution in molecular biology. This is caused by the complex nature of glycans, the difficulty to sequence them and the fact that their biosynthesis could not be linked directly to the DNA-pattern. New technologies, however, made it possible to study the structure of sugar chains. In 1988 Rademacher, Parekh and Dwerk therefore coined the term "glycobiology". Glycobiology in its broadest sense is thus the study of structure, biosynthesis and biology of saccharides that are widely distributed in nature.
3.2 PROTEOGLYCANS AND GLYCOSAMINOGLYCANS
3.2.1 Proteoglycans and glycosaminoglycans
Proteoglycans (PGs) consist of a core protein with one or several glycosaminoglycans (GAGs) attached. GAGs are linear polysaccharides, assembled of a repetitive disaccharide unit. Each disaccharide contains one amino sugar (N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc)) and one uronic acid (glucuronic acid (GlcA) or iduronic acid (IdoA)).
The existence of five GAGs is known by date, as shown in figure 3.1: hyaluronan (HA), chondroitin sulfate (CS), dermatan sulfate (DS), heparan sulfate/heparin (HS) and keratan sulfate (KS). These GAGs can be placed in two families: the glucosaminoglycans (HA and HS) and the galactosaminoglycans (CS and DS). KS is also a glucosaminoglycan, but it is in several ways a "false" glucosaminoglycan. KS contains a neutral galactose (Gal) instead of the charged hexuronic acid, can be branched and can be N-glycosidically (KS type I) or
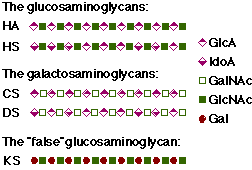
Figure 3.1 Classification of GAGs
O-glycosidically (KS type II) linked to the core protein.
Heparan sulfate (HS) and chondroitin sulfate (CS) contain a linkage sequence of four sugar units (GlcA - Gal - Gal - Xyl), which is O-glycosidically coupled to Ser or Thr in the core protein.
Different core proteins are known by date, with sizes between 14 and 580 kD (3). PGs can be divided into different families, according to their core proteins, their predominant GAG modification or according to location and function.
HSPGs can be classified into 4 families: syndecans (transmenbrane HSPGs), glypicans (bound to plasma membrane lipid by a glycosyl phosphatidylinositol linkage), perlecan and agrin (both secreted into basement membranes).
3.2.2 Heparan sulfate and heparin
3.2.2.1 The history of heparin (as reviewed in Ref. (4))
In 1918, Howell and Holt described a new anticoagulant, called heparin. Because of the used isolation procedure and the high amounts of phosphorus found in the first preparations, it was considered to be a phospholipid. In 1925 however, Howell performed a colorometric assay (Molish
a-naphtol method) which indicated the presence of carbohydrate in the purified heparin and in 1935 Jorpes proved that heparin was not a phospholipid, but a carbohydrate. The identification of the monosaccharides in heparin started in 1936, but lasted until 1964. During the same period, also other pieces of the puzzle were revealed, such as the sulfate and acetyl substituents, and in 1955 Foster and Huggard published a picture of heparin which started the discussion about the fine structure of heparin. Now we know that not all the assumptions made by Foster and Huggard were correct, but it lasted until the 1980s until reliable information about the structure of heparin was available.3.2.2.2 Structure and biosynthesis of heparin and heparan sulfate
The biosynthesis of HS starts with the enzymatic transfer of xylose (Xyl) from the sugar nucleotide to the hydroxyl group of serine residues with a specific amino-acid sequence in the core protein. Subsequently Gal, Gal and GlcA are transferred from the sugar nucleotides to the initiated linkage region by specific Golgi enzymes. Once the linkage region is formed, chain elongation starts by adding alternatingly GlcNAc and GlcA to the non-reducing end of the chain by
a
-GlcNAc transferase II and b-GlcA transferase II(5) respectively. During the heparin and heparan sulfate polymerisation in the Golgi , several enzymes modify the chain by epimerising GlcA and sulfating part of the monosaccharides (Figure 3.2 A). The Golgi enzymes involved in this modification reactions are N-deacetylase/N-sulfotransferase, uronosyl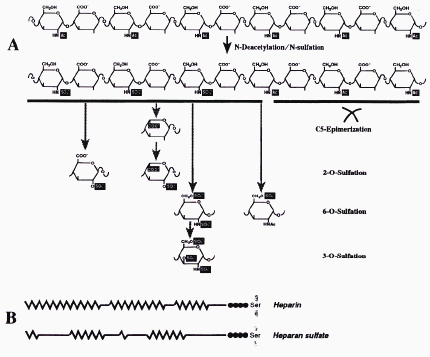
Figure 3.2 Polymer-modification reactions involved in the biosynthesis of heparin and
HS(6)
C5-epimerase, 2-O-sulfotransferase, 6-O-sulfotransferase and 3-O-sulfotransferase (reviewed in Ref. (6)).
This modifications tend to occur in clusters, as a consequence of the substrate requirements of the modifying enzymes. This results in different domains in the HS/heparin chain: the highly N-sulfated (NS) regions (presented as a zig-zag line in Figure 3.2 B), the longer and barely modified N-acetylated (NA) regions (presented as a straight line in Figure 3.2 B) and the mixed NA/NS-regions.
Considering the biosynthetic constrains, the disaccharide species given in Figure 3.3 can be expected. Species between square brackets have not been identified, but are theoretically tolerated.
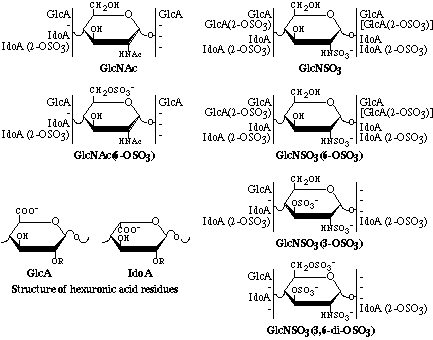
Figure 3.3 Scheme illustrating disaccharide sequences identified in heparin and HS.
Modified from Ref. (7).
3.2.2.3 The difference between heparin and heparan sulfate(5)
Heparin and heparan sulfate are produced by the same enzymes with the major difference that heparin is the most extensively modified form. It therefore is characterised by a high degree of epimerised and sulfated saccharides. A list of some important differences is shown in Table 3.1.
Table 3.1 Major differences between heparin and heparan sulfate(5).
|
Characteristic |
Heparan sulfate |
Heparin |
|
Soluble in 2 M potassium acetate |
yes |
no |
|
Size |
10-70 kD |
10-12 kD |
|
Sulfate/hexosamine |
0.8-1.8 |
1.8/2.4 |
|
GlcN N-sulfates |
40-60% |
385% |
|
IdoA content |
30-50% |
370% |
|
Binding to antithrombin |
0-0.3% |
~30% |
|
Site of synthesis |
virtually all cells |
mast cells |
|
Used as an anticoagulant |
no |
yes |
3.2.3 Functions of proteoglycans
3.2.3.1 GAG-binding proteins
Most of the physiological functions of PGs known to date are very closely related to the ability of GAG chains to bind selectively to proteins. The most important form of interaction is electrostatic interaction between the negatively charged GAG chains and the positively charged proteins, but other interactions such as hydrogen binding and hydrophobic interactions also occur in nature(6). The most famous interaction of a GAG with a protein is the binding of heparin to antithrombin. To be able to bind to antithrombin, heparin or HS chains require a specific pentasaccharide, as given in Figure 3.4. This was also the first known protein-GAG interaction with a physiological relevance (i.e. preventing the clogging of blood). By date, more then 100 GAG-binding proteins are known, of which some are given in Table 3.2, demonstrating the broad nature of these molecules. The binding of proteins to GAGs results in protein immobilisation, regulation of the enzymatic activity, binding of ligands to their receptors or protection of proteins against degradation. These interactions are of importance in haemostasis, lipid transport, lipid adsorption, cell growth, cell division and cell migration. In some of the interactions the core proteins are also important, in others the proteins just interact with the GAG. Co-operation between the core protein and the GAG chain can occur in several ways. The most simple way of co-operation is that the core protein act as a frame to hold and immobilise the GAGs. The core protein can also act as an anchor to attach the GAGs to the cell surface or to another macromolecule. A third possibility is that the core protein has a physiological function. This means that the core protein is essential to get a certain reaction between the GAG chain and the binding protein. It is for example known that the core protein of HSPGs mediates the interaction of the PG with components of the ECM, such as fibronectin, several collagens and thrombospondin (3).
Table 3.2 Some examples of GAG-binding proteins and their biological activity(8)
|
Protein |
GAG |
Physiological effect of binding |
|
|
Antithrombin |
heparin/HS |
systemic anticoagulation |
|
|
Heparin cofactor II |
DS and heparin |
localised anticoagulation |
|
|
t-Plasminogen activator |
heparin/HS |
clot dissolution |
|
|
Fibrinoblast growth factors |
heparin/HS |
mitogenesis |
|
|
Hepatocyte growth factor |
heparin/HS |
mitogenesis |
|
|
Chemokines IL-8/MIP-1b |
heparin/HS |
inflammation |
|
|
L and P selectins |
heparin/HS |
inflammation |
|
|
Extracellular superoxide dismutase |
heparin/HS |
host defence |
|
|
Lipoprotein lipase |
heparin/HS |
localised lipolysis/turnover |
|
|
apoE |
heparin/HS |
lipoprotein clearance |
|
|
Fibronectin |
heparin/HS |
cell adhesion |
|
|
Laminin |
heparin/HS |
cell adhesion |
|
|
Type V collagen |
heparin/HS |
cell adhesion |
|
|
Thrombospondin |
heparin/HS/CS |
cell adhesion/growth |
|
|
CD44 |
HA |
cell adhesion/motility |
|
|
RHAMM |
HA |
cell adhesion/motility |
|
|
Aggrecan |
HA |
cartilage formation |
|
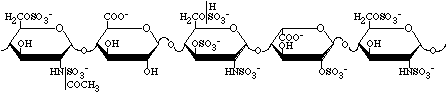
Figure 3.4 The binding region for antithrombin in heparin. Modified from Ref. (3)
3.2.3.2 The role of PGs in microbial adherence
Beside their function as an attachment site for cellular adhesion molecules and matrix molecules, in order to create stable tissue structures and extracellular matrices, cell surface PGs are also used by micro-organisms as a primary attachment site to the infected cell. Although it is known that cell surface PGs act as adhesion receptors, their precise role in invasion is indistinct. Proteoglycans mediate probably in an early stage of adherence, because heparin can block initial interaction and dislodge freshly bound organisms(2). The most important interactions between micro-organisms and proteoglycans in eukaryotic cells are given in Table 3.3.
Table 3.3 Micro-organisms bind proteoglycan receptors on eukaryotic cells(2)
|
Micro-organism |
Target tissue(s) |
||
|
Gram-negative bacteria |
|||
|
Bordetella pertussis |
Ciliated epithelium in respiratory tract |
||
|
Chlamydia trachomatis |
Eyes, genital tract, lymphoid tissues |
||
|
Helicobacter pylori |
Gastric mucosa |
||
|
Haemophilus influenzae |
Respiratory epithelium |
||
|
Borrelia burgdorfeii |
Endothelium, epithelium, ECM |
||
|
Nesseria gonorrhoeae |
Genital tract |
||
|
Gram-positive bacteria |
|||
|
Staphylococcus aureus |
Connective tissues, endothelical cells |
||
|
Streptococcus pyogenes |
Cardiac and kidney tissues |
||
|
Streptococcus mutans |
Cardiac and kidney tissues |
||
|
Streptococcus gordinii |
Cardiac tissue |
||
|
Parasites |
|||
|
Plasmodium falciparium |
Hepatocytes, placenta, endothelial cells |
||
|
Leishmania amazonensi (amastigotes) |
Macrophages, fibroblasts, epithelium |
||
|
Leishmania donovani (promastigotes) |
Macrophages, fibroblasts, epithelium |
||
|
Trypanosoma cruzi |
Heart, tract, nervous system, ECM |
||
|
Viruses |
|||
|
Dengue virus(9) |
Liver, spleen, mesenteric lymph node(10) |
||
|
HSV |
Mucosal surfaces of mouth, eyes, genital tract, respiratory tract; latent in nerve ganglia |
||
|
CMV |
Neurophils, monocytes |
||
|
HIV-1 |
T lymphocytes |
||
3.2.4 Turnover and degradation of PGs(1)
Turnover of proteoglycans can occur in two ways, either by shedding from the cell surface or by endocytosis. Shedding from the cell surface happens through proteolysis of the core protein, resulting in free extracellular glycosaminoglycans(11). The endocytic way takes place in a highly organised manner. After internalisation, the core proteins are degraded. Partial enzymatic cleavage fragments the GAG chain into molecules smaller than 10 kD. In this way, many attackpoints are created for the sequential action of exoglycosidases which cleave the chain by removing single sugar units one by one from the non-reducing ends.
3.3 STUDY OF HEPARAN SULFATE STRUCTURE AND FUNCTIONS
3.3.1 Background
Before describing the main techniques used in HS studies, I want to explain the goal of these studies. Why study the HS structure?
The goal of all the experiments is to identify the proper ligands to different proteins. Or, in other terms, to identify what can bind to a certain sequence within the HS chain. Once identified, the protein-HS interaction can be studied and also how this interactions affect the function of the protein.
In this chapter, both the main techniques used to make the samples I was working with as the main techniques I used to investigate my samples are described. The techniques I didnt use myself are only given to make the picture complete. This means that they are not described into detail. The theoretical background behind the methods I did use is only given in function of the practical work. It is certainly not a complete description.
3.3.2 Preparation of HS and modified heparin fragments
Before HS or modified heparin fragments can be used, they have to be prepared. Figure 3.5 gives an overview of the several steps used to produce these samples.
Heparin and HS can be obtained from both tissues and cell cultures. If one needs a lot of material, tissue should be used as a source. If one wants to get metabolical radiolabeling, a cell culture can be used. The properties of the collected HS species depend on the tissues used as a source. HS from bovine lung for example is different from HS from porcine liver. Cell cultures on the other hand seem to have a more uniform HS pattern.
From the cell culture or the tissue, the HS or heparin can be retrieved by quite a few extraction steps. The three main steps are removing proteins and glycoproteins, removing the core protein and removing other glycans. Each of these steps is followed by either a precipitation step or a run on an anionexchange column. As anionexchanger we have used diethylaminoethyl (DEAE) based matrices. This is a beaded cellulose ion exchanger which can be used over a wide range of molecular weights. Due to its rather weak charge, it is possible to separate long and highly sulfated chains. If one wants to get rid of CS, one must digest CS by chondroitinase and run the mixture on the DEAE column again to separate HS from the disaccharides of the CS digestion. The HS can now be characterised and used in binding assays, or one can cleave the HS chain to obtain smaller fragments.
Cleavage of HS chain can be performed in 2 major ways: chemically or enzymatically. Chemical cleavage by deamination results in a sugar change at the reducing end, where anhydromannose is formed. This structure can be reduced, so it is possible to introduce a radioactive label there. Chemical cleavage with HNO
2 at pH 1.5 is selective for N-sulfated glucosamine (GlcN). Chemical cleavage with HNO2 at pH 4.0 is selective for glucosamine with a free amino group (GlcNH2+), so it needs deacetylation first. Enzymatic cleavage is much more selective for certain sequences, and introduces a double bound at the non-reducing end of hexuronic acid.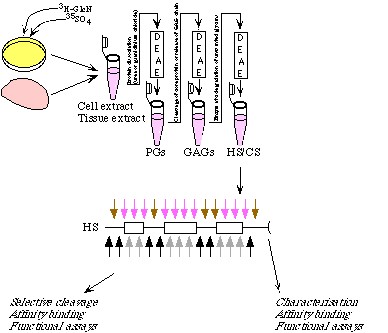
Figure 3.5 From tissue or cell to HS or modified heparin fragments
If heparin is used, it is possible to mimic the HS structure (except for the domains) by modifying heparin, but also HS can be modified to change the configuration. This is accomplished by removing and adding certain functional groups on the chain. The possible approaches are N-deacetylation or N-desulfation followed by N-sulfation or N-acetylation,
2-O-desulfation and 6-O-desulfation
Once the desired fragments are produced and radiolabeled, subfractionation is possible according to size, charge and affinity. The columns used for size and charge separation are given in Table 3.4.
Table 3.4 Columns used for size and charge separation of heparin and HS fragments
|
Size separation |
Charge separation |
|
BioGel P-10 |
DEAE |
|
Superdex 30 |
MonoQ |
|
Superose 12 |
Propac |
|
Superose 6 |
SAX-Partisil |
|
G-15 |
3.3.3 Affinity selection of heparin and HS fragments
Once heparin or HS fragments of a certain size are produced it is possible to select them based on their affinity to proteins. A scheme of this approach is given in Figure 3.6. The goal of affinity selection is to separate HS or heparin species that bind to a certain protein from those that dont bind. The theory behind the most important techniques I used for affinity selection are described in this chapter.
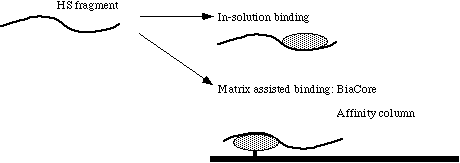 Figure 3.6 Affinity selection of HS and heparin fragments
Figure 3.6 Affinity selection of HS and heparin fragments
3.3.3.1 Phage display antibodies
Phage display antibodies are artificially made antibodies, constructed completely in vitro without using hybridoma technology or immunised animals. In this way, it is possible to make high affinity antibodies against virtually every biological molecule outside the natural host.
In nature, B lymphocytes display immunoglobulin antibodies on their surface, where they act as antigen receptors. Which antibody is displayed on the surface of a B lymphocyte depends on the arrangement of the V genes, that code the variable domain of the immunoglobulin (Ig), during the development of the B-lymphocyte. Encounters of B cells, expressing each a single type of antibody, with all the antigens present in the body, will conserve this B cell and the respective antibody.
Phage display technology imitates the immune system by rearranging the V-genes, and expressing them on the surface of a bacteriophage instead of a B-lymphocyte. This makes it possible to produce whatever combination of antibodies, also such ones that would not be produced in an organism due to tolerance towards own antigens. By screening the phages to immobilised antigens (Figure 3.7), one can separate the effective antibodies from the ineffective ones. One then infects a bacterium, such as E. coli, with the phage. Because the phage contains inside the genetic material to produce the antibody on its surface, huge amounts of soluble antibody can be made by the bacterium after infection. This process results in a phage display antibody, containing a light chain interlinked to a heavy chain, attached to a phage protein (Figure 3.8).
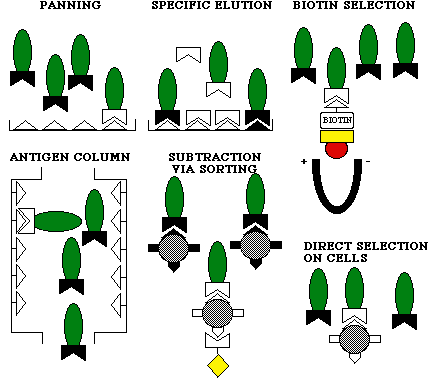
Figure 3.7 Screening of phage display antibodies to immobilised antigens(12)
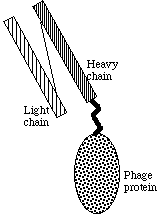
Figure 3.8 Scheme of a phage display antibody
3.3.3.2 Purification of antibodies on a Protein A Sepharose column
Protein A is an outer coat protein of Staphylococcus aureus and binds strongly with the intervariant region of immunoglobulins. Protein A also binds the phage display antibodies we used. If one immobilises protein A on a matrix (for example cross-linked agarose 4%, as in Pharmacias Protein A Sepharose), one can make a gel that binds specifically to the antibodies. If one runs a mixture of proteins through the gel, only the antibodies will remain in the gel. They can be eluted by lowering the pH after several washing steps. In this way, it is possible to purify the antibodies.
If one doesnt elute the antibodies, the gel can be used as an affinity column. This is described in 3.3.3.3.
3.3.3.3 Affinity chromatography
As shown in figure 3.9, affinity chromatography requires 3 basic tools. First of all, a matrix is needed, to which the ligand (L) can be bound (for example Protein A if an antibody is used as ligand). This can be packed into a column. If one lets a solution containing a sample flow through the column, only the samples (S) with a high affinity for the ligand will stay in the column. By eluting the samples, for example by increasing the salt concentration or changing the pH, the samples are separated based on their affinity. This method is often used to purify biomolecules, such as antibodies, receptors, DNA-binding proteins, etc.
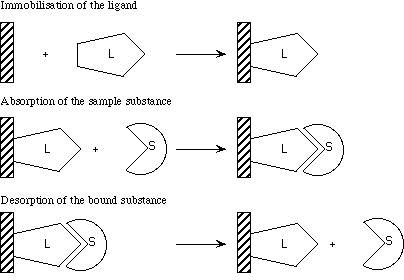
Figure 3.9 Principle of affinity chromatography (modified from Pharmacias handbook)
3.3.3.4 Biomolecular interaction analysis (BIA)
If light goes from one medium into another medium with a lower refractive index, the light will be partially reflected. Above a critical angle, depending on the refractive indices of both media and the wavelength of the light, light is totally reflected. This phenomenon is called total internal reflection (TIR). When TIR occurs, a so called evanescent wave penetrates into the medium with the lower refractive index. If the interface between the two media is covered with a thin metal film, and if the light is monochromatic and p-polarised (the electrical vector component is parallel to the plane of incidence), another phenomenon can occur, called surface plasmon resonance (SPR). This is caused by free electron clouds in the metal film, which can resonate with the evanescent wave. If this happens, a minimum can be seen in the intensity of the reflected light beam at a certain angle, as showed in figure 3.10 (nsurf is the refractive index of the bulk solution near the sensor surface). The angle of resonance changes clearly if the refractive index near the metal film changes. This makes it possible to use SPR in a biosensor. If a ligand is immobilised on the sensor surface (i.e. the metal film), binding of a soluble ligate from the bulk solution to the ligand will change the refractive index near the surface and change the resonance angle.
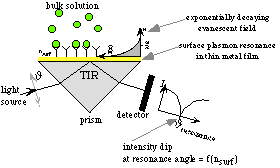
Figure 3.10 Principle of an SPR biosensor(13)
In the BIACORE
®, sensor chips are used to apply SPR in a device for real-time BIA. Ligands, which do not need to be labeled can be bound to the chip surface (as showed in figure 3.11 for biotinylated ligands), and the sensor chips can be replaced quite easily. Once a chip is made with a certain ligand, it can be used to examine several ligates.For the BIACORE
®, several chips are available with different properties. The basic principle, however, remains the same, but the immobilisation chemistry differs.
Figure 3.11 Immobilisation of a biotinylated ligand on a BIACORE
® sensor chip (modifiedfrom BIAapplications handbook)
3.3.4 Sequencing
Once a certain HS or heparin fragment is selected based on its affinity, it can be sequenced, in order to find out which saccharide sequences are responsible for the binding of the fragment to a certain protein. By date, several methods can be used, such as gel-based sequencing, mass spectrometry sequencing and sequencing of radiolabeled HS.
We used the last technique, which is based on partial cleavage of HS. By adding specific exoenzymes to the mixture of the cleavage products, some structures are digested. By comparing the results of different digestions, one can determine the sequence.
3.3.5 Theoretical background of other used techniques
3.3.5.1 SDS-PAGE by Laemmli(14)
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), is one of the most used methods for protein analysis.
Before the samples are applied, they are denaturated by cooking them with SDS in the presence of a reducing agent, such as 2-mercaptoethanol or dithiothreitol (DTT). All the proteins then have a rod-like shape and a uniform charge to mass ratio due to the SDS surrounding the proteins.
SDS-PAGE is a discontinuous system. This means that the gel buffers have a composition different from the electrode buffer or running buffer, which has a higher pH and conductivity. The samples first enter the large pore stacking gel, in which they are concentrated into small bands. The lower gel or separation gel has smaller pores and a higher pH. Here the proteins are separated by molecular weight.
The pore size of the gel is by convention given in %T. This is the weight percentage of the total acrylamide monomer including the crosslinker (bis-acrylamide) in the gel. After crosslinking the polyacrylamide, a network of pores is created in which the proteins can be separated (Figure 3.12).
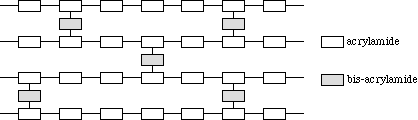
Figure 3.12 Model of polyacrylamide gel
3.3.5.2 Native PAGE
Native PAGE is based on the same principle as SDS-PAGE, but the buffers do not contain SDS or other denaturating substances. In this way, proteins are not denaturated, which makes it possible to analyse their native structure.
3.3.5.3 Ion exchange chromatography
Ion exchange chromatography is a method to separate charged molecules. The separation is based on the reversible adsorption of charged molecules in a solution to immobilised ion exchange groups of opposite charge. A typical ion exchange experiment includes 5 main stages.
The first step is the equilibration of the column. In this step, the ion exchanger is brought to a starting condition, the desired pH and ionic strength, which make it possible for charged molecules to bind. In this stage, counter-ions from the buffer are associated with the ion exchanger.
The next step is the application of the sample. While the sample flows through the column, the counter-ions are replaced by sample molecules with the appropriate charge. Sample substances that do not bind to the ion exchanger are washed away by the buffer flow.
The third step is the start of desorption. In this step, bound molecules are eluted from the column by changing the buffer conditions so that the new conditions are unfavourable for ionic adsorption of the charged molecules. A general method for doing so is to use an increasing salt gradient, in order to increase the ionic strength. By using a gradient, bound molecules are eluted in the order of their binding strengths. The most weakly bound molecules elute first, followed by the stronger bound ones.
The substances, which did not elute under previous conditions, are eluted in the fourth step. This can be accomplished by for example increasing the salt concentration even more for a certain time.
The last step is the regeneration of the column, which can be done by re-equilibrating the column with the starting buffer.
If molecules can be separated, then it is because the molecules have a different binding strength to the ion exchanger. So it is of great importance to choose the right ion exchanger in order to get a separation. If for example HS disaccharides are analysed, a strong anion exchange (SAX) column (e.g. Partisil 10) gives a good separation, while a DEAE column for example is not strong enough to separate disaccharides, but allows to discriminate between different forms of GAGs.
3.3.5.4 Western blotting
Western blotting is a method to detect very small quantities of a certain protein of interest in a sample. After electrophoresis by SDS-PAGE, the proteins are transferred to a membrane. This occurs under the influence of an electrical current and makes the proteins accessible for reaction with a monoclonal antibody against the protein of interest. This antibody can then form a complex with a secondary antibody, which is radiolabeled to be detected. Alternativerly the secondary antibody can also be detected by ELISA or by chemoluminescence. By date, also horseradish peroxidase conjugate antibodies are available. In this way one does not need a secondary antibody.
4 MATERIALS AND METHODS
4.1 Purification of phage display antibodies on protein A Sepharose beads
4.1.1 Materials
Protein A Sepharose 4B-CL (Pharmacia 17-0780-01)
Periplasmatic fractions containing phage display antibodies (received from T. van Kuppevelt, University of Nijmegen, NL)
Binding buffer: Tris buffered saline, sterile filtered (TBS: 50 mM Tris pH 7.5 - 150 mM NaCl)
Elution buffer: 0.1 M glycine pH 2.5, sterile filtered
Neutralisation buffer: 1 M Tris, sterile filtered
Storage buffer: TBS - 0.05 % NaN
pH paper
Shaking board
Ice
Labtop centrifuge
Poly-Prep Column (BioRad 731-1550)
Radiolabeled heparin or HS preparations
4.1.2 Procedure
Protein A Sepharose was prepared freshly by suspending it in distilled water and washing it with approximately 200 ml of distilled water, or reused after desorption of bound material.
The beads were washed three times with 10 ml TBS and centrifuged for 5 minutes at 300 rpm. The buffer was removed with a suction pump.
2 ml of periplasmatic phage extract was added to approximately 1 ml of beads.
The beads were incubated for 3 hours at room temperature, or overnight at 4 ûC under slight shaking.
The gel was centrifuged again as above.
The supernatant was removed, marked and stored at -20 ûC.
10 ml TBS was added to the beads, and they were shaken softly for about 5 minutes.
The gels were centrifuged again and this washing step was repeated 5 times totally.
The Protein A Sepharose was transferred to the BioRad Poly-Prep Column, and the tubes were rinsed out to prevent loss of beads.
The antibodies were eluted by applying 6 x 500 µl portions of 0.1M glycine pH 2.5.
Each portion was collected as a separate fraction in an Eppendorf tube, the pH was checked with pH paper and neutralised with 1M Tris if necessary. The fractions were kept on ice all the time.
The amount of proteins in each tube was determined using the BCA method (miniscale at 60 ûC) and positive fractions were stored as aliquots at -20 ûC after addition of BSA to 1 mg/ml.
Positive fractions were analysed by SDS-PAGE on a 12 % gel, together with the supernatant and the protein A Sepharose was transferred back into 10 ml tubes and kept in storage buffer.
In the same way antibody columns were prepared, with the only difference that the antibodies were not eluted when the beads were transferred into the BioRad column. These antibody columns were used for affinity chromatography.
Affinity chromatography
500 µl of the solution containing the desired amount of HS or heparin preparations was incubated on the closed antibody column for 30 minutes on ice.
After incubation, the GAGs were eluted with ice cold TBS containing different salt concentrations to obtain an affinity selection. The elution buffers used for analytical and preparative affinity chromatography are given in Table 4.1.
Table 4.1 Elution of GAGs from antibody columns
|
Analytical affinity chromatography |
Preparative affinity chromatography |
|
5 x 1 ml TBS (150 mM NaCl) |
7 x 1 ml TBS (150 mM NaCl) |
|
5 x 1 ml TBS (200 mM NaCl) |
7 x 1 ml TBS (200 mM NaCl) |
|
5 x 1 ml TBS (400 mM NaCl) |
5 x 1 ml TBS (400 mM NaCl) |
|
3 x 1 ml TBS (600 mM NaCl) |
3 x 1 ml TBS (600 mM NaCl) |
|
3 x 1 ml TBS (2 M NaCl) |
3 x 1 ml TBS (2 M NaCl) |
Each fraction was collected in a scintillation vial. When performing an analytical run, the entire eluent was mixed with 2 ml scintillation cocktail and counted in the scintillation counter. For preparative runs, a few µl was transferred to another vial, mixed with 1 ml of scintillation cocktail and counted.
4.2 Protein determination by bca
4.2.1 Materials
Reagent A:
1 % BCA-Na2
2 % Na2CO3·H2O
0.16 % Na2-tartrate
0.4 % NaOH
0.95 % NaHCO3
pH 11.25 by adding 50% NaOH
Reagent B:
4% CuSO4·5 H2O
BSA 1 mg/ml standard
Glycine-Tris buffer (50 vol. 0.1 M glycine pH 2.5 - 3 vol. Tris 1M)
Thermoblock
ELISA plate
ELISA plate reader with 562 nm filter
4.2.2 Procedure (15)
100 volumes of reagent A were mixed with 2 volumes of reagent B to obtain the working reagent.
A BSA standard dilution series was made as in Table 4.2.
Table 4.2 Dilution series of BSA
|
BSA 1 mg/ml µl |
glycine-Tris buffer (50:3) µl |
H 2Oµl |
|
0 |
25 |
25 |
|
5 |
25 |
20 |
|
10 |
25 |
15 |
|
15 |
25 |
10 |
|
20 |
25 |
5 |
|
25 |
25 |
0 |
0-25 µl sample was mixed with 25 µl H2O and glycine-Tris (50:3) until 50 µl.
50 µl sample or standard were mixed with 1 ml working reagent and incubated at 60 ûC for 30 minutes.
After cooling down, 100 µl of each sample was pipetted into an ELISA plate and the absorbance was measured at 562 nm.
4.3 nitrocellulose-trapping assay for heparin-binding proteins(16)
4.3.1 Materials
Round nitrocellulose (NC) filters, pore size 0.45 µm (Sartorius)
Filtration device to apply sample under vacuum
1 x PBS-CMF (phosphate buffered saline, Ca and Mg free: 8.2 g NaCl - 0.2 g KCl - 1.44 g Na2HPO4·2 H2O - 0.2 g KH2PO4 - H2O
2 x PBS-CMF (phosphate buffered saline, Ca and Mg free: 16.4 g NaCl - 0.4 g KCl - 2.88 g Na2HPO4·2H2O - 0.4 g KH2PO4 - H2O
Æ 1 l) 2 M NaCl
H2O
Heparin binding protein
BSA 10 mg/ml
Radiolabeled GAGs, diluted in H2O
Scintillation cocktail (Wallac, OptiPhase HiSafe 3, containing poly(ethyleneglycol)mono(4-nonylphenyl)-ether as a hazardous component)
Shaking board
4.3.2 Procedure
The protein was dissolved in 2 x PBS-CMF - 0.2 mg/ml BSA at the wished concentration.
100 µl of the protein dilution was put in an Eppendorf tube, and mixed with 100 µl of a suitable GAG dilution.
100 µl of the same GAG dilution was put directly into a scintillation vial (input).
The content of the Eppendorf tube was mixed and incubated for 1 hour at room temperature.
The filters were prepared on the suction device and washed 2 times with 5 ml PBS-CMF.
Samples were applied while the vacuum was on.
The filters were washed 2 times with 5 ml of the same buffer used for incubation.
The filters were removed and transferred into 20 ml scintillation vials.
2 ml 2M NaCl was added to the vials and the GAGs were eluted by shaking 15-30 minutes on a shaking board.
10 ml of scintillation cocktail was added and the tubes were shaken well.
The input vials were treated the same way to have identical solvent conditions.
The samples were counted in a scintillation counter.
4.4 SDS-PAGE
4.4.1 Materials
Gel preparation and electrophoresis
4 x Upper gel buffer (0.5 M Tris - 0.4% SDS - HCl
4 x Lower gel buffer (1.5 M Tris - 0.4% SDS - HCl
Æ pH 8.8 - H2O Æ 1 l) 5 x Running buffer (15.15 g Tris - 72.05 g glycine - 25 ml of 20 % SDS - H2O
Æ 1 l) Acrylamide stock (300 g acrylamide - 8 g methylenebisacrylamide - H2O
Æ 1 l) Sucrose stock (30%)
4 x sample solution (-) (16 ml 0.5 M Tris-HCl pH 8.0 - 20.52 g sucrose - 4 ml 0.1 M Na2EDTA - 40 ml 20 % SDS - H2O
Æ 92 ml) 20 % (m/v) SDS
10 % (m/v) ammonium persulfate (APS)
TEMED (N,N,NN-Tetramethylethyleneamine)
0.5 M dithiothreitol (DTT)
0.1 % bromophenolblue (BPB)
Overlay: butanol, saturated with 1 x lower gel buffer
Mini-gel device (BioRad Mini-PROTEAN® 3 Cell, 165-3301)
Electrophoresis power supply
Coomassie staining
Staining solution (0.25 % Coomassie Brilliant Blue R - 1 % CH3COOH - 50 % CH3OH - H2O
Æ 1 l) Destaining solution (1 % CH3COOH - 45 % CH3OH - H2O
Æ 1 l) Glycerol solution (2 % in distilled water)
Cellophane
Drying frame
Silver staining
Fixing solution (40 % ethanol - 10 % acetic acid - 50% H2O)
Incubation solution (75 ml ethanol - 17.0 g sodium acetate·3H2O - 1.3 glutardialdehyde (25 % w/v) - 0.50 g sodium thiosulfate·5H2O - H2O
Æ 250 ml) Silver solution (0.25 g silver nitrate - 50 µl formaldehyde - H2O
Æ 250 ml) Developing solution (6.25 g sodium carbonate - 25 µl formaldehyde - H2O
Æ 250 ml) Stop solution (3.56 g Na2EDTA·2H2O - H2O
Æ 250 ml) Glycerol solution (10% in distilled water)
4.4.2 Procedure
Gel preparation and electrophoresis(14)
The glass plates were rinsed well.
A spacer plate and a short plate were selected according to the gel thickness (0.75 mm), assembled and placed in the casting frame.
The casting frame was put into the casting stand and the lower gel with the desired percentage was prepared as in Table 4.3.
Table 4.3 SDS-PAGE lower gel
|
5 |
6 |
7.5 |
10 |
12 |
15 |
% |
|
|
4 x lower gel buffer |
1.25 |
1.25 |
1.25 |
1.25 |
1.25 |
1.25 |
ml |
|
acrylamide (30:.8) |
0.835 |
1.0 |
1.25 |
1.665 |
2.0 |
2.5 |
ml |
|
H2O |
2.915 |
2.75 |
2.5 |
2.085 |
1.75 |
1.25 |
ml |
|
APS (10 %) |
0.025 |
0.025 |
0.025 |
0.025 |
0.025 |
0.025 |
ml |
|
TEMED |
0.005 |
0.005 |
0.005 |
0.005 |
0.005 |
0.005 |
ml |
The gel was poured between the glass plates, covered with overlay and left to polymerise.
After polymerisation, the butanol was removed and washed away with distilled water and the upper gel was prepared by mixing 500 µl 4 x upper gel buffer, 200 µl acrylamide (300:8), 1.3 ml sucrose 30%, 10 µl APS and 2µl TEMED.
The upper gel was poured on top of the lower gel and the comb was applied.
Samples were prepared by mixing 15 µl sample with 4.6 µl 4 x sample solution (-) and cooking it for 5 minutes at 96 ûC. After cooling down, the samples were centrifuged and mixed with 0.4 µl DTT:BPB (1:1).
The samples were applied and the system was connected to the power supply. During migration through the upper gel, 120 V was applied. In the lower gel 200 V was applied.
After the electrophoresis, the upper gel was removed and the lower gel was stained by Coomassie staining or silver staining.
Coomassie staining
The gel was stained with the staining solution for 30 minutes.
After staining, the gel was washed with distilled water and placed in destaining solution for several hours or several days until the protein bands were clearly visible.
After destaining, the gel was placed in 2% glycerol for 20 minutes.
The upper gel was then removed and the lower gel was placed between two layers of cellophane, placed in the drying frame and dried.
Silver staining (17)
The gel was put in fixing solution first, in which it was slightly shaken for 30 minutes.
To wash away the acetic acid, the gel was incubated for 30 minutes, or overnight, in incubation solution.
After 3 times washing with distilled water for 5 minutes, the gel was treated with silver solution for 40 minutes.
To visualise the bands, the gel was slightly shaken in developing solution for 15 minutes.
The development was stopped after 15 minutes, or earlier if the bands were dark enough, by washing the gels 2 times for 5 till 10 minutes in stop solution.
After 3 times washing with distilled water for 5 minutes, the gel was put in glycerol solution for 20 minutes and dried between cellophane
4.5 NATIVE PAGE
4.5.1 Materials
4 x Upper gel buffer (0.5 M Tris - HCl
4 x Lower gel buffer (1.5 M Tris - HCl
Æ pH 8.8 - H2O Æ 1 l) 5 x Running buffer (15.15 g Tris - 72.05 g glycine - H2O
Æ 1 l) Acrylamide stock (300 g acrylamide - 8 g methylenebisacrylamide - H2O
Æ 1 l) Sucrose stock (30%)
4 x sample solution (-) (16 ml 0.5 M Tris-HCl pH 8.0 - 20.52 g sucrose - 4 ml 0.1 M Na2EDTA - H2O
Æ 92 ml) 10 % (m/v) ammonium persulfate (APS)
TEMED (N,N,NN-Tetramethylethyleneamine)
0.1 % bromophenolblue (BPB)
Overlay: butanol, saturated with 1 x lower gel buffer
Mini-gel device (BioRad Mini-PROTEAN® 3 Cell, 165-3301)
Electrophoresis power supply
4.5.2 Procedure
The glass plates were rinsed well.
A spacer plate and a short plate were selected according to the gel thickness (0.75 mm), assembled and placed in the casting frame.
The casting frame was put into the casting stand and the lower gel with the desired percentage was prepared as in Table 4.4.
Table 4.4 Native PAGE lower gel
|
5 |
6 |
7.5 |
10 |
12 |
15 |
% |
|
|
4 x lower gel buffer |
1.25 |
1.25 |
1.25 |
1.25 |
1.25 |
1.25 |
ml |
|
acrylamide (30:.8) |
0.835 |
1.0 |
1.25 |
1.665 |
2.0 |
2.5 |
ml |
|
H2O |
2.915 |
2.75 |
2.5 |
2.085 |
1.75 |
1.25 |
ml |
|
APS (10 %) |
0.025 |
0.025 |
0.025 |
0.025 |
0.025 |
0.025 |
ml |
|
TEMED |
0.005 |
0.005 |
0.005 |
0.005 |
0.005 |
0.005 |
ml |
The gel was poured between the glass plates, covered with overlay and polymerised for 60 minutes.
After polymerising, the butanol was removed and washed away with distilled water and the upper gel was prepared by mixing 500 µl 4 x upper gel buffer, 200 µl acrylamide (300:8), 1.3 ml sucrose 30%, 10 µl APS and 2µl TEMED.
The upper gel was poured on top of the lower gel and the comb was applied.
Samples were prepared by mixing 15 µl sample with 4.6 µl 4 x sample solution (-).
After cooling down, the samples were centrifuged and mixed with 0.4 µl BPB.
The samples were applied and the system was connected to the power supply. During migration through the upper gel, 120 V was applied. In the lower gel 200 V was applied.
After the electrophoresis, the upper gel was removed and the lower gel was stained by Coomassie staining or silver staining.
4.6 TRIS-BORATE POLYACRYLAMIDE GEL FOR ANIONIC POLYSACCHARIDES(18)
4.6.1 Materials
Acrylamide stock (300 g acrylamide - 8 g methylenebisacrylamide - H2O
10 x running/gel buffer stock (10 x TBE: 108 g Tris - 55 g boric acid - 7.4 g Na2EDTA - H2O
Æ 1 l) 10 x sample buffer (2 M sucrose in 1 x running buffer)
10 % (m/v) ammonium persulfate (APS)
TEMED (N,N,NN-Tetramethylethyleneamine)
Dye standard: 0.02 % phenol red
Mini-gel device (BioRad Mini-PROTEAN® 3 Cell, 165-3301)
Electrophoresis power supply
Alcian blue solution (0.5 % alcian blue in 25 % isopropanol - 3% acetic acid - 72 % distilled water)
Destaining solution (25 % isopropanol - 3% acetic acid - 72 % distilled water)
Glycerol solution (2 % in distilled water)
Cellophane
Drying frame
4.6.2 Procedure
The glass plates were rinsed well.
A spacer plate and a short plate were selected according to the gel thickness (0.75 mm), assembled and placed in the casting frame.
The casting frame was put into the casting stand and the lower gel with the desired percentage was prepared according to Table 4.4
Table 4.4 Tris-borate PAGE gel
|
5 |
7.5 |
10 |
12.5 |
15 |
17.5 |
20 |
25 |
% |
|
|
10 x gel buffer |
1.6 |
1.6 |
1.6 |
1.6 |
1.6 |
1.6 |
1.6 |
1.6 |
ml |
|
acrylamide (30:.8) |
2.6 |
3.9 |
5.2 |
6.5 |
7.8 |
9.1 |
10.4 |
13.0 |
ml |
|
H2O |
11.75 |
10.45 |
9.15 |
7.85 |
6.55 |
5.25 |
3.95 |
1.35 |
ml |
|
APS (10 %) |
60 |
60 |
60 |
60 |
60 |
60 |
60 |
60 |
µl |
|
TEMED |
6 |
6 |
6 |
6 |
6 |
6 |
6 |
6 |
µl |
The gel was left to polymerise for 1 hour.
The samples were prepared by diluting them in 1 x sample buffer and adding the dye standard.
The gel was pre-electrophoreses for 1 hour at 120 V.
After applying the samples submarine with a Hamilton syringe, the gel was run at 120 V for approximately 1 hour.
The gel was stained in alcian blue solution and destained in destaining solution.
The gel was incubated 20 minutes in glycerol solution and dried between cellophane.
4.7 URONIC ACID DETERMINATION(19)
4.7.1 Materials
25 mM sodium tetraborate in concentrated H2SO4
0.15 % phenylphenol (meta-hydroxy-diphenyl) in 0.5 % NaOH
1 mg/ml glucuronic acid standard in water
Ice
Water bath
4.7.2 Procedure
Samples were diluted in distilled water so that 50 µl of the sample contained between 0 and 50 µg of carbohydrate.
A dilution series of the standard was made by diluting 0, 5, 10, 15, 20 and 25 µl glucuronic acid in distilled water to a total volume of 50 µl.
Both standards and samples were kept on ice while adding 300 µl of ice-cold H2SO4-borate.
After 5 minutes, the samples and standards were boiled for 5 minutes in a water bath of 100 ûC.
After cooling, 5 µl of phenylphenol reagent was added and vortexed immediately.
After 5 minutes, the absorbance was read at 520 nm.
4.8 BIOMOLECULAR INTERACTION ANALYSIS OF HEPARIN AND HEPARAN SULFATE FRAGMENTS ON THE BIACORE
4.8.1 Materials
BIACORE 2000
PIONEER Sensor Chip B1 or CM5
HEPES buffered saline (HBS: 10 mM HEPES, 150 mM NaCl, 3mM EDTA)
EDC (N-ethyl-N-(3-dimethyl aminopropyl)-carbodiimide hydrochloride) (BIACORE Amine Coupling Kit BR-1000-50)
NHS (N-hydroxysuccinimide) (BIACORE Amine Coupling Kit BR-1000-50)
Streptavidin solution (20 µl 1.5 mg/ml + 80 µl 10 mM Ac- pH 5)
Ethanolamine hydrochloride (1 M in water, pH 8.5)
Antibody solution in HBS-CMD (HBS + 1 mg/ml carboxymethyl dextran)
4.8.2 Procedure (20)
Immobilisation of biotinylated heparin or HS fragments on a B1 or CM5 chip
The flow rate for the continuous flow buffer (HBS) was programmed (20 µl/min)
300 µl EDC/NHS (0.2 M / 0.05 M) was injected at the same flow rate and the signal before and after the injection was written down.
120 µl streptavidin was injected, still at the same flow rate, and the signal before and after the injection was written down.
300 µl ethanolamine hydrochloride solution was injected and the signal before and after the injection was written down.
120 µl biotinylated heparin(fragments) were injected, and the chip was washed with HBS.
Kinetic measurements of interactions between soluble antibodies and immobilised heparin or HS fragments
When using manual operation in the BIACORE Control Software, a manual sensorgram was started as described in the BIACORE 2000 Instrument Handbook.
After setting the detection mode and the flow path, the flow rate was set and the sample was injected.
After the completion of the sample injection, the sensorgram was stored and evaluated with the BiaEvaluation software.
When using method-controlled operation, a method program was written in the BIACORE Method Definition Language, as described in Appendiced E and F of the BIACORE 2000 Instrument Handbook. Results are automatically saved and can also be evaluated with the BiaEvaluation software.
4.9 CHARGE SEPARATION OF HS AND HEPARIN FRAGMENTS ON A MONO-Q
COLUMN
4.9.1 Materials
HPLC equipment
MonoQ column (Amersham Pharmacia Biotech)
Buffer A: 50 mM Na-acetate, sterile filtered
Buffer B: 50 mM Na-acetate - 2 M NaCl, sterile filtered
Radiolabeled heparin and HS fragments
Fraction collector
4.9.2 Procedure
First a small amount of sample was injected to establish the ideal salt gradient.
Once the best gradient was found, the samples were injected and run with the same gradient.
Of each run, the eluted fractions were collected in a fraction collector and each fraction was counted in a scintillation counter.
4.10 WESTERN BLOTTING
4.10.1 Materials
SDS-PAGE gel
Transfer buffer (25 mM Tris - 192 mM glycin - 20% Methanol)
Whatman 3 MM filter paper
polyvinylidene difluoride (PVDF) membrane
Trans-Blot SD Semi-Dry Transfer Cell (BioRad 170-3940)
Amido black (0.1% in 45% methanol)
Methanol
Blocking buffer (PBS - 15% ficoll calf serum - 0.1% Tween20)
Monoclonal antibody, horseradish peroxidase conjugate (Invitrogen 951-25)
Washing buffer (PBS - 0.1% Tween20)
Detection solution (ECL+, amersham pharmacia biotech RPN 2132)
Photographic film
4.10.2 Procedure
The PVDF membrane was wetted with methanol first and then with transfer buffer.
The SDS-PAGE gel was put on top of a PVDF membrane between 2 times 3 sheets of filter paper of about the same size as the gel, wetted with transfer buffer.
The proteins were then transferred onto the membrane by applying 0.8 mA / cm2 for 1 hour.
After transfer the gel was stained with amido black or specifically stained with a monoclonal, horseradish peroxidase conjugated, antibody.
For specific staining, the membrane was blocked by shaking gently in blocking buffer for 1 hour.
The antibody (when using a horseradish conjugate, no secundary antibody has to be used) was added to the blocking buffer membrane so that a 5000 fold dilution of the antibody was obtained. The membrane was incubated in the antibody solution for 1 hour.
The membrane was then washed in washing buffer.
After washing, the membrane was removed and the excess of washing buffer was drained.
The membrane was placed protein side up on a sheet of kitchen foil and covered with detection solution.
After 5 minutes, the excess of detection solution was temoved by holding the membrane in forceps and touching the edge agains a tissue.
The membrane was wrapped in a fresh sheet of kitchen foil and exposed on a photographic film for 15 seconds.
5 RESULTS AND DISCUSSION
5.1 PURIFICATION OF THE ANTIBODY
5.1.1 Determination of the antibody concentration
The antibodies were purified on a protein A column and eluted. After the elution, the protein concentration in each elutent was measured using the BCA method. Due to the rather low protein concentrations, the values are situated near the detection limit. In that way, the values give only an idea of the real protein concentration. The obtained results, with the 95% confidence intervals, are:
276 µg (± 276 µg) in 1 ml of antibody EV3C3 periplasmatic fraction (27.05.99)
368 µg (± 129 µg) in 2 ml of antibody EV3C3 periplasmatic fraction (27.05.99)
103 µg (± 41 µg) in 2 ml of antibody EV3C3 periplasmatic fraction (17.02.00)
5.1.2 Control of the antibody purity
5.1.2.1 SDS-PAGE
The purified antibody (EV3C3, 17.02.00) was electrophoresed on an SDS-polyacrylamidegel, together with the original periplasmatic fraction and the supernatant. The results of this gel is shown in Figure 5.1. Lane one contains the periplasmatic fraction, lane two the supernatant and lane three the purified antibody.

Figure 5.1 SDS-PAGE of the purified antibody with the supernatant and the original
periplasmatic fraction
After SDS-PAGE, two of the prepared gels were used for Western blotting. One of the gels was just transfered to a membrane and stained with amidoblack (Figure 5.2). The other one was blotted with a secondary antibody and developped (Figure 5.3).

Figure 5.2 Amido black stained proteins on a PVDF membrane

Figure 5.3 Blotted antibody
5.1.2.1 Native PAGE
The purified antibody (EV3C3, 17.02.00) was electrophoresed on an SDS-polyacrylamidegel, together with the original periplasmatic fraction and the supernatant. The results of this gel is shown in Figure 5.4.
(NATIVE GEL)
Figure 5.4 Native PAGE of the purified antibody with the supernatant and the original
periplasmatic fraction
After SDS-PAGE, two of the prepared gels were used for Western blotting. One of the gels was just transfered to a membrane and stained with amidoblack (Figure 5.5). The other one was blotted with a secondary antibody and developped (Figure 5.6).

Figure 5.5 Amido black stained membrane
Figure 5.6
5.2 DETERMINATION OF THE BINDING PROPERTIES BETWEEN THE
ANTIBODY AND HEPARIN OR HS
5.2.1 Nitrocellulose-trapping assay
5.2.1.1 Experimental data
for the first experiment a dilution series of 3H-heparin from bovine lung was made and icubated with antibody EV3C3
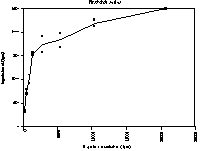
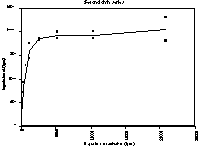
Figure 5.2 In solution binding of antibody EV3C3 with 3H-labeled heparin (bovine lung)
5.2.1.2 Data analysis
The data from the in solution binding assay were analised both by linear (Scatchard plot) and non-linear regression in order to get a Kd value for the binding between heparin from bovine lung and antibody EV3C3. The obtained values are:
For linear regression: Kd = ~11 nM
For non-linear regression: Kd = 12.0 nM ± 2.7 nM
Although it is known that linear regression is not as reliable as nonlinear regression, we used linearisation to be able to compare the data from non-linear regression.
5.2.2 Affinitychromatography of HS and heparin preparations
For the affinitychromatography assays I used HS preparation #12 pools VII and VIII. This preparation is received from Akzo Nobel, where it is a byproduct in the heparin production. The HS was cleaved by deacetylation with HNO2 at pH 3.9 for 15 minutes. It was labeled with NaB3H4. Pool VII and VIII contain aproximately octamers.
An overview of the obtained results is given in figure 5.3. The first graph gives the distribution of the original preparation on an antibody EV3C3 column. Approximately 10.5 % is situated in the "bound fraction", which elutes with 400 mM NaCl. The distribution of the eluent after re-application of both the 150mM and the 400 mM NaCl eluent are given in graphs
a and b. Graphs g, d and e show the distribution of several fraction on a MonoQ-column.5.2.1 Biomolecular interaction analysis on the BIACORE

Figure 5.3 Affinitychromatography of HS preparation #12 pool VII & VIII on an antibody
EV3C3 column
6 CONCLUDING REMARKS
7 REFERENCES
1. Freeze, H., Degradation and Turnover of Glycans, in Essentials of Glycobiology, A. Varki, et al., Editor. 1999, Cold Spring Harbor Laboratory Press: New York. p. 267.
2. Rostand, K.S. and Esko, J.D. (1997)Infection and Immunity65, 1
3. Kjellén, L. and Lindahl, U. (1991)Annu. Rev. Biochem.60, 443
4. Rodén, L., Highlights in the history of heparin, in Heparin, D. Lane and U. Lindahl, Editor. 1989, Edward Arnold: London. p. 1.
5. Esko, J., Proteoglycans and Glycosaminoglycans, in Essentials of Glycobiology, A. Varki, et al., Editor. 1999, Cold Spring Harbor Laboratory Press: New York. p. 145.
6. Lindahl, U., Lidholt, K., Spillmann, D., and Kjellén, L. (1994)Thromb. Res.75, 1
7. Lindahl, U., Biosynthesis of heparin and related polysaccharides, in Heparin - chemical and biological properties, clinical applications, D.A. Lane and U. Lindahl, Editor. 1989, Edward Arnold: London. p. 159.
8. Esko, J., Glycosaminoglycan-binding Proteins, in Essentilas of Glycobiology, A. Varki, et al., Editor. 1999, Cold Spring Harbor Laboratory Press: New York. p. 441.
9. Chen, Y., Maguire, T., Hileman, R., Fromm, J., Esko, J., Linhardt, R., and Marks, R. (1997)Nature Medicin3, 828
10. Rosen, L., Drouet, M., and Deubel, V. (1999)Am. J. Trop. Med. Hyg.61, 720
11. Bernfield, M., Götte, M., Park, W., Reizes, O., Fitzgerald, M., Lincecum, J., and Zako, M. (1999)Annu. Rev. Biochem.68, 729
12. Hoogenboom, H. (1997)TIBTECH15, 62
13. Schuck, P. (1997)Annual Review of Biophysics and Biomolecular Structure26, 541
14. Laemmli, U.K. (1970)Nature227, 680
15. Smith, P., et al. (1985)Anal. Biochem.150, 76
16. Maccarana, M. and Lindahl, U. (1993)Glycobiology3, 271
17. Heukeshoven, J. and Dernick, R. (1985)Electrophoresis6, 103
18. Pelkonen, S. and Finne, J. (1989)Methods Enzymol.179, 104
19. Blumenkrantz, N. and Asboe-Hansen, G. (1973)Anal. Biochem.54, 484
20. BIACORE 2000 Instrument Handbook. Biacore AB. 1999, Uppsala