APOLIPOPROTEIN
B
Apoliprotein
B (107 mg/dl):
This is the protein cap that each LDL particle wears. Over 90% of
low density lipoprotein (LDL) particle is composed of Apo
B. It serves the function of solubalizing cholesterol within the LDL
complex, which in turn increases
the
transport capacity of LDL for subsequent deposit on the arterial wall.
Apo B is therefore a convenient marker for assessing the cholesterol depositing
capacity of the blood, and studies have clearly indicated it as a better
discriminator of angiographically documented coronary artery disease than
LDL cholesterol. By counting these, you get a precise measure of the LDL
particles in the bloodstream, a truer indication of your genetic predisposition
to heart disease. These particles may damage your arteries and cause
blockages, so it helps to know how many youve got. This is based
on the following studies:
"Effects
of apolipoprotein and low density lipoprotein receptor gene polymorphisms
on lipid metabolism, and the lipid risk factors of coronary artery disease."
Apo
B exists in human plasma as two isoforms, apo B-48 and apo B-100. Apo B-100
is the major physiological ligand for the LDL receptor. It is the largest
monomeric protein sequenced so far, containing 4536 amino acid residues
(Chen et al. 1986, Law et al. 1986). Its gene has been mapped on the short
arm of chromosome 2, with an approximate length of 43 kilobases and 29
exons
(Ludwig
et al. 1987). The LDL-binding domain of the molecule is proposed to be
located between the residues 3129 and 3532 (Knott et al. 1986). Apo B-100
is synthesised in the liver and is required for the assembly of very low
density lipoproteins (VLDL). It does not interchange between lipoprotein
particles, as do the other lipoproteins, and it is found in IDL and LDL
particles after the removal of the apolipoproteins A, E and C (Young 1990).
Apo
B-48 is present in chylomicrons and chylomicron remnants and plays an essential
role in the intestinal absorption of dietary fats (Kane 1983). Apo B-48
is synthesised in the small intestine. It comprises the N-terminal 48%
of apo B-100 and is produced due to posttransscriptional apo B-100 mRNA
editing at codon 2153, which creates a stop codon in the intestine instead
of a glutamine in the liver (Chen et al. 1987).
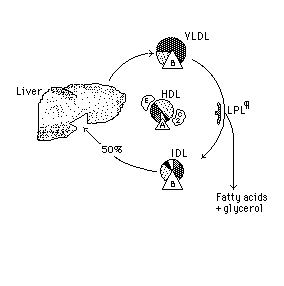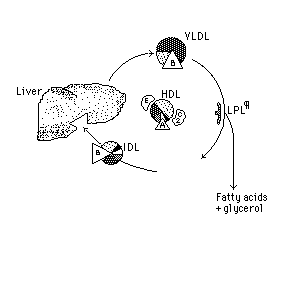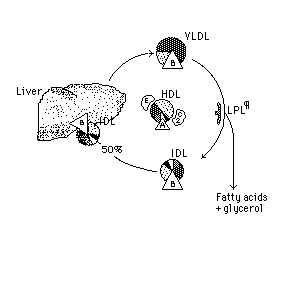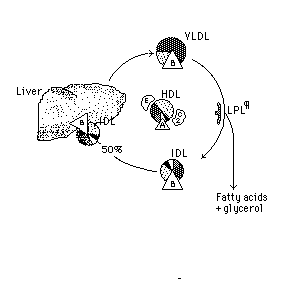
The
triglyceride of the other 50% of the IDL is hydrolyzed by another enzyme
*hepatic lipase* producing LDL, a lipoprotein that is
richer
than IDL in cholesterol and its esters. (Show this sequence again.)
Lipoprotein
Interconversions
The
whole sequence of lipoprotein interconversions is as follows:
Apo
B is incorporated into VLDL by hepatocytes, preparing this lipoprotein
for transport of triglycerides (TG) and cholesterol from liver to other
tissues. The VLDL as it is secreted is in an incomplete *nascent* state.
The conversion of nascent VLDL to its functional form needs the addition
of Apo E and Apo C2 donated by *HDL* which in turn acquires them from other
lipoproteins to form the "mature" *VLDL* that transports lipids.
When
VLDL encounters lipoprotein lipase *LPL* in tissue capillaries the Apo
C2 on the VLDL activates the enzyme, which hydrolyzes much of the triglyceride
of the VLDL to produce *IDL*. The latter releases Apo C2 and Apo E which
are recycled to HDL and then to VLDL.
About
1/2 of the resulting IDL, which is poorer than VLDL in triglyceride and
relatively richer in cholesterol and its esters, is taken up by the liver
by a receptor that recognizes the *Apo B* of the IDL. The *LDL* is taken
up by the liver with its Apo B acting as the ligand to the receptor. Another
24% of the LDL is delivered to *other tissues* leaving about 1% of the
LDL to be removed from the circulation by scavenger cells such as those
found in atheromatous plaques.
However,
the receptors on scavenger cells do not recognize "native" Apo B. Rather,
they have a specific affinity for *oxidized* Apo B which is formed when
LDL persists for an abnormally long time in the circulation.
Persistence
of LDL in the circulation may result from the excessive VLDL production
associated with a high dietary intake of fats, especially those rich in
saturated fatty acids. It also occurs in primary and secondary lipidemias
in which there is a subnormal uptake of IDL and LDL by the liver and other
tissues.
Mutations
Mutations
occurring in the apo B gene can alter blood cholesterol levels. Most of
the mutations lower blood cholesterol levels due to the production of truncated
apo B. The mechanisms by which blood cholesterol is lowered are not yet
fully understood. Two mutations in the apo B gene have been associated
with elevated blood cholesterol. The apo B-3500 ArgÆGln substitution
causes
familial
defective hypercholesterolemia (FDB) due to defective binding of LDL to
its receptor (Vega & Grundy 1986, Soria et al. 1989). The prevalence
of the mutation in the general population in Central Europe is 1/204-1/700
(Innerarity et al.1990, Tybjaerg-Hansen et al. 1990, Schuster et al. 1990).
The highest prevalence has been reported from Switzerland (Miserez et al.
1994), and so far apo B -3500 has not been found in Finland (Hämäläinen
et al. 1990). Another mutation in the LDL receptor binding area causing
apo B-3531 ArgÆ Cys has been described to cause moderate hypercholesterolemia
due to defective binding of LDL to its receptor (Pullinger et al. 1995).
Several
restriction fragment length polymorphisms (RFLP) in the Apo B gene have
been defined (Humphries & Talmud 1995). The most widely studied of
these is the XbaI polymorphism in exon 26, which does not result in an
amino acid substitution. In some populations the presence of the XbaI cutting
site is associated with hypercholesterolemia in both normolipemic (Berg
1986, Talmud et al. 1987, Aalto-Setälä et al. 1988) and hypercholesterolemic
(Leren et al. 1988, Aalto-Setälä et al. 1989) individuals. The
absence of the XbaI cutting site was associated with higher triglyceride
levels in one study (Deeb et al. 1986). Several studies have failed to
reveal any association between the XbaI polymorphism and lipid values (Hegele
et al. 1986, Aburatani et al. 1988,
Rajput-Williams et al. 1988, Darnfors et al. 1989, Gajra et al. 1994,)
and in one study the association of the presence of the XbaI cutting site
with elevated cholesterol and triglyceride levels was only observed in
patients with peripheral artery disease (Monsalve et al. 1988).
The
EcoRI restriction fragment length polymorphism in exon 29 is associated
with an amino acid change Gln Æ Lys4154 . Most studies have revealed
no association between the EcoRI polymorphism and cholesterol or triglyceride
levels (Ma et al. 1987, Dunning et al. 1988, Jenner et al. 1988, Aburatani
et al. 1988, Peacock et al. 1992,). An association between elevated triglycerides
and the absence of the EcoRI cutting site has been reported in coronary
heart disease patients (Paulweber et al. 1990, Tybjaerg-Hansen et al. 1991)
and in healthy males (Paulweber et al. 1990).
The
MspI RFLP in exon 26 is associated with an amino acid change Arg Æ
Gln3611. The MspI polymorphism is not associated with differences in serum
lipid concentrations (Deeb et al. 1986, Hegele et al. 1986, Xu et al. 1989,
Genest et al. 1990).
The
apo B signal peptide contains a leucine-alanine-leucine insertion/deletion
polymorphism affecting the amino acids 14-16 producing signal peptides
with 24 or 27 amino acids (Boerwinkle & Chan 1989). The ins allele
has been associated with elevated serum triglycerides (Tikkanen & Heliö
1992), low serum cholesterol and apo B (Hansen et al. 1993), and coronary
artery disease
(Peacock
et al. 1992) in some populations, whereas in others the del allele has
been connected with elevated total and LDL cholesterol but not with myocardial
infarction (Bohn et al. 1994). No association between the polymorphism
and lipids was detected in Asian patients, but the del allele was associated
with coronary artery disease (Wu et al. 1994). A strong linkage disequilibrium
between
the XbaI and ins/del polymorphisms has been reported (Hansen et al. 1993).
RECOMBINANT
ADENO-ASSOCIATED VIRUS-MEDIATED GENE DELIVERY OF APOLIPOPROTEIN B mRNA
SITE-SPECIFIC RIBOZYME (Abstract No. 430)
Shihua
Sun, Talesha Ford, Alan Davis, Ba-Bie Teng
Research
Center for Human Genetics, Institute of Molecular Medicine, University
of Texas-Houston, Houston, TX
Center
for Gene Therapy, Baylor College of Medicine, Houston, TX
Abstract
Apolipoprotein
B (apoB) plays an obligatory role in the production of triglyceride-rich
lipoprotein particles and it is necessary
for
the transport of lipids and nutrients in the circulation. However, overproduction
of apoB is strongly associated with
premature
coronary artery diseases. Patients with familial hypercholesterolemia have
markedly elevated plasma levels of
cholesterol
and apoB and develop atherosclerosis. To modulate apoB production, we designed
a hammerhead ribozyme
targeted
at GUA6679Ø of apoB mRNA (designated RB15) to cleave apoB mRNA in
vivo. From our previous study, we used
E1-deleted
adenovirus vector to deliver RB15 to a dyslipidemia mouse model. The study
showed that RB15 cleaved apoB
mRNA
efficiently. There was a marked reduction of apoB gene expression and decrease
plasma levels of cholesterol,
triglyceride,
and human apoB100. Therefore, apoB mRNA-specific hammerhead ribozyme can
be used as a potential
therapeutic
agent to modulate apoB gene expression and to treat hyperlipidemia.
To have a long-term gene expression, no immune response, and no toxicity
in gene therapy, in this study, we sought to construct liver-specific adeno-associated
virus (AAV) vector to deliver RB15 to HepG2 cells and animals. RB15 is
driven by transthyretin liver-specific promoter (TTR) and a 2773-bp human
genomic fragment of hypoxanthine guanine phosphoribosyltransferase (HPRT)
was inserted downstream of 5 ITR of AAV vector (pAAV-TTR-RB15). We produced
rAAV-TTR-RB15 by co-transfection of pAAV-TTR-RB15 with helper plasmid pDG
in 293 cells, followed by purification using non-ionic iodixanol gradient
and by ion exchange with heparin affinity chromatography. The virus titer
was 1 x 1012 particles/ml, determined by both real-time PCR and dot-blot
hybridization. We characterized the rAAV virial capsid proteins (VP1, VP2
and VP3) by western blotting.
The
rAAV-TTR-RB15 (1 x 108 particles) was used to infect HepG2 cells. Total
RNA was extracted at days 3 and 7 after infection. Using RNase protection
assay the levels of apoB mRNA on day 7 was barely detectable (7.6% compared
to that of
non-treated
samples). The rAAV-TTR-RB15 (8 x 1010 particles) was used to transduce
mouse overexpressing human apoB
gene.
Using both PCR and real-time PCR RB15 DNA was detectable in the mouse liver
on day 45 after treatment. The RB15
RNA
was also detected in mouse liver on day 45 after treatment by RT/PCR. Southern
blot analysis show that rAAV-TTR-RB15 was stably transduced into the liver.
Using Western blot analysis, the levels of human apoB decreased on days
7, 14, and 28 to 13%, 40%, and 63%, respectively, compared to that of day
0 before treatment. In conclusion, the expressed ribozyme RB15 RNA was
active, which decreased apoB production.

|
This is
about ApoB.
(See the whole
sequence of lipoprotein
interconversions.)
Clicking
on an *asterisked*
word below takes you to that apoprotein.
The
triglyceride of the other 50% of the IDL is hydrolyzed by another enzyme
*hepatic lipase*
producing LDL, a lipoprotein that is richer than IDL in cholesterol and
its esters.
-------------------------------------
See the whole
sequence of lipoprotein
interconversions or click on the individual
steps below again.
Apo
B is incorporated into VLDL by hepatocytes, preparing this lipoprotein
for transport of triglycerides (TG) and cholesterol from liver to other
tissues. The VLDL as it is secreted is in an incomplete *nascent*
state.
The conversion of nascent VLDL to its functional form needs the addition
of Apo E and Apo C2 donated by *HDL*
which in turn acquires them from other lipoproteins to form the "mature"
*VLDL*
that transports lipids.
×××××
When VLDL encounters lipoprotein lipase *LPL*
in tissue capillaries the Apo C2 on the VLDL activates the enzyme, which
hydrolyzes much of the triglyceride of the VLDL to produce *IDL*.
The latter releases Apo C2 and Apo E which are recycled to HDL and then
to VLDL.
×××××
About 1/2 of the resulting IDL, which is poorer than VLDL in triglyceride
and relatively richer in cholesterol and its esters, is taken up by the
liver by a receptor that recognizes the *Apo
B* of the IDL.
The
*LDL* is taken up by the liver with its Apo B acting as the ligand to the
receptor. Another 24% of the LDL is delivered to *other
tissues* leaving about 1% of the LDL
to be removed from the circulation by scavenger cells such as those found
in atheromatous plaques.
×××××
However, the receptors on scavenger cells do not recognize "native" Apo
B. Rather, they have a specific affinity for *oxidized*
Apo B which is formed when LDL persists for an abnormally long time in
the circulation.
×××××
Persistence of LDL in the circulation may result from the excessive VLDL
production associated with a high dietary intake of fats, especially those
rich in saturated fatty acids. It also occurs in primary and secondary
lipidemias in which there is a subnormal uptake of IDL and LDL by the liver
and other tissues.
×××××
|
Fasting
Insulin and Apolipoprotein B Levels and Low-Density Lipoprotein Particle
Size as Risk Factors for Ischemic Heart Disease
Benoît
Lamarche, PhD; André Tchernof, PhD; Pascale Mauriège, PhD;
Bernard Cantin, MD; Gilles R. Dagenais, MD; Paul J. Lupien,
MD;
Jean-Pierre Després, PhD
Context.
Epidemiological studies have established a relationship between cholesterol
and low-density lipoprotein cholesterol (LDL-C)
concentrations
and the risk of ischemic heart disease (IHD), but up to half of patients
with IHD may have cholesterol levels in the normal range.
Objective.
To assess the ability to predict the risk of IHD using a cluster of nontraditional
metabolic risk factors that includes elevated fasting insulin and apolipoprotein
B levels as well as small, dense LDL particles.
Design.
Nested case-control study.
Setting.
Cases and controls were identified from the population-based cohort of
the Québec Cardiovascular Study, a prospective study conducted in
men free of IHD in 1985 and followed up for 5 years.
Participants.
Incident IHD cases were matched with controls selected from among the sample
of men who remained IHD free
during
follow-up. Matching variables were age, smoking habits, body mass index,
and alcohol consumption. The sample included 85
complete
pairs of nondiabetic IHD cases and controls.
Main
Outcome Measures. Ability of fasting insulin level, apolipoprotein
B level, and LDL particle diameter to predict IHD events, defined as angina,
coronary insufficiency, nonfatal myocardial infarction, and coronary death.
Results.
The risk of IHD was significantly increased in men who had elevated fasting
plasma insulin and apolipoprotein B levels and
small,
dense LDL particles, compared with men who had normal levels for 2 of these
3 risk factors (odds ratio [OR], 5.9; 95%
confidence
interval [CI], 2.3-15.4). Multivariate adjustment for LDL-C, triglycerides,
and high-density lipoprotein cholesterol (HDL-C) did not attenuate the
relationship between the cluster of nontraditional risk factors and IHD
(OR, 5.2; 95% CI, 1.7-15.7). On the other hand, the risk of IHD in men
having a combination of elevated LDL-C and triglyceride levels and reduced
HDL-C levels was no longer significant (OR, 1.4; 95% CI, 0.5-3.5) after
multivariate adjustment for fasting plasma insulin level, apolipoprotein
B level, and LDL particle size.
Conclusion.
Results from this prospective study suggest that the measurement of fasting
plasma insulin level, apolipoprotein B level,
and
LDL particle size may provide further information on the risk of IHD compared
with the information provided by conventional lipid
variables.
JAMA.
1998;279:1955-1961
OVER
THE LAST 30 years, several epidemiological studies have reported a direct
relationship between total plasma cholesterol and
low-density
lipoprotein cholesterol (LDL-C) concentrations and the risk of coronary
artery disease (CAD), and elevated total plasma cholesterol levels are
considered by many to be the main cause of coronary atherosclerosis. However,
the ability to adequately identify individuals at high risk for the development
of CAD solely on the basis of total cholesterol or LDL-C concentration
has recently been challenged by evidence suggesting that a considerable
proportion of patients with CAD may have cholesterol levels in the normal
range (Genest et al reported the proportion to be as high as 50%).
There are also data to suggest that a notable proportion of patients undergoing
cholesterol-lowering therapy and who achieve significant LDL-C reduction
may still develop CAD. These observations have emphasized the need to find
additional markers of risk that would allow a more refined identification
of individuals at high risk for CAD.
Clinical
data have provided evidence that elevated plasma triglyceride levels and
reduced high-density lipoprotein cholesterol (HDL-C)
concentrations
may be associated with a considerable increase in CAD risk. Although the
independent contribution of plasma
triglycerides
to CAD remains controversial, the clinical relevance of elevated triglyceride
levels should no longer be overlooked as
hypertriglyceridemia
may reflect additional metabolic disturbances highly predictive of CAD
risk. Results from the Helsinki Heart Study and from the Prospective
Cardiovascular Münster (PROCAM) study have suggested that hypertriglyceridemia
should be considered an important risk factor for CAD, particularly when
combined with elevated LDL-C and reduced HDL-C concentrations. This cluster
of risk factors may represent the metabolic condition most predictive of
CAD risk.
With
the 5-year prospective data from the Québec Cardiovascular Study,
we have recently reported that elevated fasting plasma insulin levels,11
elevated apolipoprotein B concentrations, and the presence of small, dense
LDL particles14 were strongly associated with the development of ischemic
heart disease (IHD) in men, independent of established risk factors. Plasma
LDL-C, triglyceride, and HDL-C levels were also significant correlates
of IHD in the Québec Cardiovascular Study. In the current study,
we investigate whether the ability to identify individuals at high risk
for the development of IHD could be improved by measuring 3 nontraditional
risk factors, namely fasting plasma insulin and apolipoprotein B levels
and LDL particle diameter, over and beyond what can be achieved using more
traditional lipid risk factors, triglyceride, LDL-C, and HDL-C levels.
METHODS
Study
Population and Follow-up
The
Québec Cardiovascular Study cohort has been described in detail
previously. In 1973, a random sample of 4637 men aged 35 to 64 years
was recruited from 7 suburbs of the Québec metropolitan area for
an evaluation of cardiovascular risk factors using the provincial electoral
lists. Subsequent evaluations were performed at regular intervals and data
collected in 1985 were used as the baseline characteristics for the present
prospective analyses. In 1985, 2443 (61%) of the living cohort came to
the lipid clinic in a fasting state for their evaluation. Among the 1557
other potential living subjects, 150 (10%) could not be located, 302 (19%)
came to the clinic in a nonfasting state, and 1105 (71%) either refused
to participate or were evaluated in a nonfasting state at their home by
project nurses. Analyses of data collected in 1973 revealed that
the age distribution of the 2443 subjects in 1985 was representative of
the original cohort. At the end of follow-up (September 1, 1990),
all subjects were contacted by mail and invited to answer a short standardized
questionnaire on smoking habits, medication use, history of cardiovascular
disease, and diabetes mellitus. For those who reported such diseases and
those who died, hospital charts were reviewed. Telephone calls were
made to subjects who did not answer a second letter and if the call was
unsuccessful, another call was made to a close family member. Mortality
and morbidity data were obtained in 99% and 96%, respectively, of the subjects
of the initial 1973 screening.
Evaluation
of Risk Factors
Data
on demographic and lifestyle variables as well as medical history and medication
were obtained in 1985 through a standardized
questionnaire
administered to each subject by trained nurses and further reviewed by
a physician. Body weight and height were
recorded.
Resting blood pressure was measured after a 5-minute rest
in
a sitting position. The mean of 2 blood pressure measures taken 5
minutes apart was used in the analyses. Information on personal and
family
history of IHD and diabetes mellitus, smoking habits, alcohol
consumption,
and medication use was also obtained. Diabetes
mellitus
was considered in men who self-reported the disease or who
were
treated with hypoglycemic agents. Only 2% of men were using
hypolipidemic
drugs in 1985 (mainly clofibrate and cholestyramine),
whereas
8% and 4% of men were using -blockers and diuretics,
respectively,
on a regular basis at the 1985 screening. Data on drug
use
at the time of follow-up were not available. Alcohol consumption
was
computed from the type of beverage (beer, wine, or spirits)
consumed
in ounces per week and then standardized as an absolute
quantity
(1 oz of absolute alcohol was equivalent to 22.5 g of alcoholic
beverage). Family history of IHD was considered positive if at least 1
parent
or 1 sibling had a history of IHD.
Definition
of IHD Events
The
diagnosis of a first IHD event included typical effort angina,
coronary
insufficiency, nonfatal myocardial infarction, and coronary
death.
All myocardial infarction cases met the criteria previously
described,16
namely diagnostic electrocardiographic (ECG) changes
alone
or 2 of the following criteria: typical chest pain of at least 20
minutes
in duration, creatine kinase enzyme level at least twice the
upper
limit of normal, or characteristic ECG changes. Coronary
insufficiency
was considered if typical retrosternal chest pain of at
least 15 minutes in duration was associated with transient ischemic
ECG
changes but without significant elevation in levels of creatine
kinase.
Diagnoses of myocardial infarction and coronary insufficiency
were
confirmed by hospital charts. All ECG tracings were read by the
same
cardiologist, who was unaware of the subjects' risk profiles. The
diagnosis
of effort angina was based on typical symptoms of
retrosternal
squeezing or pressure-type discomfort occurring on
exertion
and relieved by rest and/or nitroglycerine. Criteria for the
diagnosis
of coronary death included confirmation from death
certificate
or autopsy report confirming the presence of coronary
disease
without evidence for noncardiac disease that could explain
death.
Myocardial infarction was considered fatal if death occurred
within 4 weeks of the initial event or if it was diagnosed at autopsy.
Deaths
related to IHD were confirmed from the Provincial Death
Registry.
Informed consent was obtained to review relevant hospital
files.
Autopsies were performed in about one third of deaths. The total
IHD
event frequency during the 5-year follow-up period was similar in
men
participating in the study (5.4%) and in nonparticipants (6.5%).
Pairing
Procedures
Between
1985 and 1990, 114 of the 2103 men who had no clinical
evidence
of IHD at baseline had a first IHD event: 50 had a myocardial
infarction,
40 had effort angina, 9 had coronary insufficiency, and 15
died
of IHD-related causes. Each case subject was matched with a
control
subject selected from among the remaining 1989 men without
IHD
during follow-up. Subjects were matched on the basis of age,
cigarette smoking, body mass index, and weekly alcohol intake. The
mean difference within pairs was 0.6 years, 0.2 kg/m2, and 0.2 oz/wk
for
age, body mass index, and alcohol intake, respectively. The mean
difference
within pairs for cigarette smoking was 0.3 cigarettes per
day.
Subjects who had an IHD event and who were classified as
nonsmokers
were systematically matched with nonsmoking
control-group
subjects.
Laboratory
Analyses
Fasting
lipoprotein lipid and apolipoprotein levels were measured in
plasma
in 1985 when subjects came to the clinic for evaluation.
Aliquots
of fasting plasma were frozen at the time of collection and
were later used for the assessment of LDL diameter and fasting
insulin concentrations. Total cholesterol and triglyceride levels were
determined on a multianalyzer (Technicon RA-500, Bayer Corp,
Tarrytown,
NY) as previously described.17 High-density lipoprotein
cholesterol
was measured in the supernatant fraction after
precipitation
of apolipoprotein Bcontaining lipoproteins with
heparinmanganese
chloride.18 Low-density lipoprotein cholesterol
levels were estimated by the equation of Friedewald et al.19 Subjects
with
triglyceride levels higher than 4.5 mmol/L (399 mg/dL) (n=52)
were excluded from the analyses.13 Plasma apolipoprotein B levels
were measured by the rocket immunoelectrophoresis method of
Laurell,20
as described previously.17 Serum standards for the
apolipoprotein
assay were prepared in the laboratory and calibrated
against
serum samples from the Centers for Disease Control and
Prevention.
The standards were lyophilized and stored at-85°C until
use.
The coefficients of variation for cholesterol, HDL-C, triglyceride,
and
apolipoprotein B measurements were less than 3%.
Low-density
lipoprotein particle diameter was assessed using
nondenaturing
2% to 16% polyacrylamide gradient gel electrophoresis
of
whole plasma according to Krauss et al21 and McNamara et al,22
as
described previously.23 Plasma samples were applied on gels in a
final
concentration of 20% sucrose and 0.25% bromophenol blue.
Following a 15-minute pre-run, electrophoresis was performed at 200
V
for 12 to 16 hours and at 400 V for 2 to 4 hours. Gels were stained
with
Sudan black B according to standardized procedures and stored
in a solution of 9% acetic acid and 20% methanol until analysis using
an
optical densitometric image analyzer (BioImage Visage
1101DGEL,
Genomic Solutions, Ann Arbor, Mich) coupled with a
computer (SPARC Station 2 Sun, Genomic Solutions). Low-density
lipoprotein diameter was estimated by comparing the migration
distance on the gel of the predominant LDL subspecies for each
individual with the migration distance of standards of known
diameters.
One assay was performed for each subject. Analyses of
pooled plasma standards revealed that the assessment of LDL
diameter
using this method was highly reproducible with a coefficient
of variation of less than 3% (A.T., unpublished data, 1996).
Fasting
plasma insulin concentrations were measured with a
commercial double-antibody radioimmunoassay (human
insulin-specific
radioimmunoassay method; Linco Research, St Louis,
Mo)
according to the manufacturer protocol. This assay shows
essentially
no cross-reactivity with human proinsulin (<0.2%). The
coefficient
of variation was below 5.5% for both low and high fasting
insulin
concentrations.11
Statistical
Analyses
Fasting insulin levels and LDL diameter were measured in 106 and
103 case-control pairs, respectively,11, 14 but data for both variables
were available simultaneously in 100 controls and 102 cases. Men
who reported having diabetes mellitus or who were receiving
hypoglycemic therapy at the baseline evaluation were excluded (15
cases and 1 control). We therefore had data on 87 IHD cases and 99
controls. After excluding all pairs for which 1 of the 2 subjects had
missing data, the study sample included 85 complete pairs of IHD
cases and matched controls. Baseline characteristics of subjects
who developed IHD during the 5-year follow-up (IHD cases) were
compared with the characteristics of those who remained IHD free
using paired t tests for means and 2 tests for frequency data.
Variables with a skewed distribution were log-transformed. Correlation
analyses were performed using the Pearson and the Spearman
coefficients of correlation for parametric and nonparametric variables,
respectively.
The median of the control group was used as the cutoff point to
identify men with elevated or low levels of each variable of interest
(LDL-C, 3.7 mmol/L [143 mg/dL]; triglycerides, 1.52 mmol/L [135
mg/dL]; apolipoprotein B, 1.1 g/L [110 mg/dL]; fasting insulin, 72
pmol/L [10 µU/mL]; HDL-C, 1.01 mmol/L [39 mg/dL]; LDL particle
diameter, 25.82 nm). Thus, by definition, each of these risk factors
was found in 50% of the control subjects. The proportion of cases
classified as having 1 or more risk factor based on these arbitrary
cutoff points was compared with that of control subjects. The
proportional hazards regression (PHREG) procedure on SAS (SAS
Institute, Cary, NC) for conditional logistic regression analysis was
used to estimate the odds ratio (OR) for IHD associated with the
presence of each risk factor, as an isolated condition or combined
with others. Odds ratios were adjusted for medication use at baseline
(-blockers and/or diuretics), family history, and systolic blood
pressure. The potential confounding effects of using -blockers and
diuretics were combined because they both yielded similar risk. Thus,
medication use (yes or no) and family history (presence or absence)
were treated as categoric variables whereas systolic blood pressure
was treated as continuous.
RESULTS
Table 1 presents the clinical characteristics of the 85 controls and
IHD cases. A higher proportion of case patients was using -blockers
and/or diuretics on a regular basis at baseline (17.7% vs 4.7%,
P=.007). However, there was no difference between cases and
controls in the use of hypolipidemic medication at baseline. As a
result of the matching procedure, the frequency of smokers (41%) and
the number of cigarettes smoked per day (25 cigarettes per day) were
essentially the same in both groups. Systolic blood pressure was
also the same in both groups. As expected, there were marked
differences in several plasma lipoprotein-lipid parameters as well as in
fasting insulin levels at baseline between IHD cases and controls.
Triglycerides (18.2%), fasting insulin (18.9%), and apolipoprotein B
(15.9%) levels showed the largest case-control differences. Mean
plasma HDL-C concentrations and LDL diameter were also
significantly different between cases and controls (P=.03). It is
important to note that although being tightly matched with IHD cases
on the basis of age, body mass index, smoking, and alcohol
consumption, the risk profile of control subjects in the current study
is
very similar to that of the total sample of men who remained free of
IHD during follow-up13 and from which they were selected.
Prevalence of Lipoprotein and Insulin Abnormalities
Because there are currently no reference values for apolipoprotein B
and insulin levels and for LDL diameter, and in an attempt to compare
the contribution to IHD risk of variables having different scales,
lipoprotein-lipid and fasting insulin levels were dichotomized using the
median (50th percentile) of the control group. Table 2 presents the
prevalence of each of the metabolic abnormalities in IHD cases.
Based on these prevalences, ORs for developing IHD during the
5-year follow-up were estimated using conditional logistic regression
while taking into consideration the potential confounding effects of
systolic blood pressure, medication use, and family history of IHD.
Eighty-one percent of cases had elevated fasting insulin
concentrations based on these criteria, yielding a 5.5-fold increase in
the OR for IHD (95% confidence interval [CI], 2.3-13.6, P<.001)
compared with men having insulin levels below the 50th percentile of
controls. Elevated plasma triglyceride levels were also associated
with a marked increase in the risk of IHD (OR, 3.5; 95% CI, 1.6-7.4;
P=.002). Elevated apolipoprotein B and LDL-C levels and small,
dense LDL particles were observed in a similar proportion of cases
(69.4%, 68.2%, and 69.4%, respectively). These 3 abnormalities were
associated with a significant 2.4-fold to 2.7-fold increase in the OR for
IHD. Finally, 62.4% of IHD cases had HDL-C levels below the 50th
percentile of controls. There was a 60% increase in the risk of IHD
associated with reduced HDL-C levels (OR, 1.6), which was not
significant after adjustment for confounders (95% CI, 0.85-3.0). This
analysis did not take into consideration the fact that cases with 1
abnormality may also have had additional metabolic abnormalities in
combination. Nevertheless, results presented in Table 2 suggest that
among all variables of interest, elevated fasting plasma insulin
concentrations, irrespective of the presence or absence of other
lipoprotein abnormalities, were associated with the greatest relative
increase in the risk of IHD.
Prevalence of Isolated Abnormalities
The prevalence rates of elevated plasma fasting insulin and
apolipoprotein B levels as well as of small, dense LDL in their isolated
form (ie, associated with none of the other 2 abnormalities) were low
in both IHD cases and control subjects. Isolated hyperinsulinemia
was observed in only 11 (12.9%) of both IHD cases and controls.
However, when considering only subjects with elevated fasting insulin
levels (42 controls and 69 cases), 11 (15.9%) of 69 hyperinsulinemic
IHD cases did not have elevated apolipoprotein B levels or small,
dense LDL in combination compared with 11 (26.6%) of 42 controls.
Only 2 (2.4%) of 85 IHD cases had isolated elevations in
apolipoprotein B levels compared with 9 (10.6%) of 85 controls.
Finally, the small, dense LDL phenotype in its isolated form was
found in only 5 (5.9%) of 85 IHD cases. In comparison, twice as many
controls (11 [12.9%] of 85) had small, dense LDL in isolation. These
results suggest that hyperinsulinemia, elevated apolipoprotein B
levels, and small, dense LDL particles may be observed more
frequently in combination with each other rather than as isolated
conditions, and that a smaller proportion of IHD cases may display
these abnormalities in their isolated form compared with controls. We
therefore tested whether the cluster of these metabolic risk factors
may further increase the risk of IHD.
Prevalence of Nontraditional Risk Factors
Figure 1 compares the prevalence rates of the cumulative number of
abnormalities in IHD cases and controls. To simplify data
presentation, fasting plasma insulin levels, apolipoprotein B levels,
and small, dense LDL particles are referred to as nontraditional risk
factors, whereas LDL-C, triglyceride, and HDL-C levels are referred to
as traditional risk factors. As shown in Figure 1 (top), only 2 IHD
cases (2.4%) had none of the 3 nontraditional metabolic risk factors,
compared with 14 controls (16.5%). One of every 5 IHD cases (n =
18, 21.2%) had 1 of the nontraditional risk factors in its isolated form,
compared with more than a third of controls (n = 31, 36.5%). The
proportion of cases that simultaneously had elevated fasting insulin
levels, elevated apolipoprotein B levels, and small, dense LDL
particles (cumulative number of risk factors, 3) was 2.6-fold greater
than that of controls (45.8% vs 17.7%). Consequently, 98% of IHD
cases had at least 1 of the nontraditional risk factors compared with
83% of controls. On the other hand, 82% of controls did not have
elevated fasting plasma insulin levels, elevated apolipoprotein B
levels, and small, dense LDL simultaneously, compared with 54% of
IHD cases.
Prevalence of Traditional Risk Factors
A similar analysis was performed using the traditional risk factors
(LDL-C, triglycerides, and HDL-C levels) as discriminating variables for
the determination of IHD risk (Figure 1, bottom). Although differences
in the proportion of cumulative number of traditional risk factors
between IHD cases and controls were slightly attenuated compared
with differences in the proportion of nontraditional risk factors, a
similar pattern of distribution was observed. There was a greater
proportion of controls that had relatively low LDL-C and triglyceride
levels and high HDL-C levels (number of risk factors, 0) compared with
IHD cases (18.8% vs 7.1%), whereas the proportion of IHD cases that
had elevated LDL-C and triglyceride levels and low HDL-C
concentrations simultaneously (cumulative number of risk factors, 3)
was 1.9-fold greater than that of controls (41.2% vs 21.2%).
Risk of Developing IHD During Follow-up
Based on the prevalence of the cumulative number of risk factors
presented in Figure 1, the crude OR for developing IHD during the
5-year follow-up was increased 18.2-fold in subjects who had all 3
nontraditional risk factors simultaneously compared with those who
had none of the 3 risk factors (results not shown). By comparison, the
OR for IHD in subjects with the 3 traditional risk factors
simultaneously was 5.2 (not shown). Multivariate conditional logistic
regression analysis was performed to compare the ability to predict
IHD using traditional and nontraditional risk factors. The prevalence of
IHD cases in subjects with no risk factor (2 and 6 IHD cases for
nontraditional and traditional risk factors, respectively) was too small
to accurately assess the risk of IHD using this group as a reference.
We have therefore performed the multivariate logistic regression
analysis by combining subjects with 0 and 1 risk factor only, and by
using this group as a reference (OR, 1). As shown in Table 3,
subjects that had elevated LDL-C and triglyceride levels and reduced
HDL-C concentrations simultaneously (cumulative number of
traditional risk factors, 3) showed a 3-fold increase in the risk of IHD
(model 1: OR, 3.0; 95% CI, 1.4-6.4; P=.005) compared with men
having none or only 1 of these risk factors. This increased risk was no
longer significant after multivariate adjustment for fasting insulin and
apolipoprotein B levels and LDL particle diameter (model 2: OR, 1.4;
95% CI, 0.5-3.5; P=.50).
The impact of having elevated fasting insulin and apolipoprotein B
levels and small, dense LDL particles in combination with each other
on the odds of developing IHD was more prominent. The risk of
developing IHD was increased almost 6-fold when subjects
simultaneously had elevated fasting insulin and apolipoprotein B
levels and small, dense LDL particles (model 3: OR, 5.9; 95% CI,
2.3-15.4; P<.001). This increase in risk was essentially unmodified
when LDL-C, triglyceride, and HDL-C levels were included as
confounders in the multivariate logistic regression model (model 4:
OR, 5.2; 95% CI, 1.7-15.7; P=.003).
An analysis was carried out to test the 2-way and 3-way interaction
terms as predictors of IHD risk. It was found that none of the 2-way or
3-way interaction terms for continuous variables were significant.
However, because of the small sample size, the possibility of a
significant interaction among the 3 nontraditional or the 3 traditional
risk factors cannot be excluded.
Univariate associations between the traditional and nontraditional risk
factors and the variables that were used to match IHD cases to
controls were investigated. Plasma triglyceride levels (r=0.15, P=.05)
and HDL-C levels (r=-0.17, P=.02) showed significant associations
with body mass index. Plasma triglyceride levels also showed a
significant but inverse correlation with age (r=-0.23, P=.003) whereas
HDL-C levels were positively associated with weekly alcohol
consumption (r=0.26, P<.001). Low-density lipoprotein particle size
was also a significant correlate of age (r=0.19, P=.01) but the most
significant correlation between risk factors and matching variables
was observed between plasma fasting insulin concentrations and
body mass index (r=0.40, P<.001).
COMMENT
Results of the present prospective study emphasize the potential of
plasma fasting insulin and apolipoprotein B levels as well as of small,
dense LDL particles as clinically relevant markers of the risk of
developing IHD. Our results suggest that this cluster of metabolic
abnormalities may even provide more information on IHD risk than the
more traditional lipid risk factors, LDL-C, triglycerides, and HDL-C.
Indeed, almost 1 (45.8%) of every 2 IHD cases had elevated insulin
and apolipoprotein B levels as well as small, dense LDL particles, and
this combination of metabolic risk factors resulted in a remarkable
18-fold increase in the risk of IHD. Adjustment for the more traditional
cluster of risk factors through multivariate logistic regression did not
attenuate this relationship. These observations have consequential
clinical implications, particularly in terms of primary prevention of IHD.
They imply that identification of individuals at risk could be
substantially improved by measuring fasting plasma insulin and
apolipoprotein B levels and LDL particle diameter. It should be kept in
mind that these findings do not in any way lessen the clinical
importance of assessing LDL-C, triglyceride, and HDL-C
concentrations. The current study should not be considered an
attempt to discredit the well-described and accepted relationship
between the so-called lipid triad and the risk of IHD.8-10 It was
apparent that an important proportion of IHD cases was characterized
by this dyslipidemia compared with controls.
It may be argued that the paired nature of the study population may
have had the adverse effect of overmatching for the traditional risk
factors, thereby understating their true impact on a randomly selected
population. As expected, there were significant correlations between
risk factors and some of the variables used to match IHD cases and
controls. Although significant, these correlations were of very low
magnitude (with shared variances lower than 7%), with the exception
of the relationship between plasma fasting insulin levels and body
mass index (with a shared variance of 16%). The paired nature of the
study is therefore very unlikely to have biased the estimation of the
contribution of the traditional risk factors to IHD risk compared with
that of the nontraditional risk factors.
We reported that a very small proportion of IHD cases had no risk
factor and that abnormalities in insulin and apolipoprotein B levels and
in LDL particle diameter were more frequently observed in
combination and not in isolation compared with controls. It is
therefore apparent that the risk of developing IHD is largely dependent
on the presence of risk factors that, in most cases, emerge as a
cluster of metabolic abnormalities. In this context, arguments have
been proposed for why plasma insulin and apolipoprotein B levels and
LDL particle size may represent better markers of IHD risk than
LDL-C, triglyceride, and HDL-C levels.
Small, Dense LDL and the Risk of IHD
Plasma LDL-C levels are merely measurements of the cholesterol
content of a lipoprotein particle that has been described as being very
heterogeneous in terms of composition, size, and density. Although
the cholesterol content of LDL certainly contributes to its
heterogeneity, we have failed to find a significant association between
LDL density or size and LDL-C levels.14, 23 Recognition of the
atherogenic potential of small, dense LDL largely came from
cross-sectional case-control studies that reported a higher prevalence
of small, dense LDL in patients with IHD compared with healthy
controls.24-26 Observations from 3 recent prospective reports provided
further support for a critical role of small, dense LDL particles in the
etiology of atherosclerosis.14, 27, 28 The greater susceptibility of these
particles to oxidation29 and their reduced affinity for the hepatic LDL
receptor30 have been proposed as potential mechanisms for the
increased atherogenic potential of small, dense LDL.
Apolipoprotein B and the Risk of IHD
Apolipoprotein B is the protein moiety of LDL. The clinical interest of
this protein lies in the fact that it provides a relatively accurate
estimate of circulating LDL particle numbers. Total plasma
apolipoprotein B concentration, as opposed to LDL apolipoprotein B,
also accounts for the number of triglyceride-rich lipoproteins (very
low-density lipoprotein and intermediate-density lipoproteins), and
recent data suggest that these 2 lipoprotein subfractions may also
play an important role in the etiology of IHD.31, 32 Plasma
apolipoprotein B concentration can therefore be considered a crude
marker of the number of atherogenic particles in plasma.33 Results
from the Québec Cardiovascular Study suggest that plasma
apolipoprotein B concentration is a strong predictor of IHD risk,
independent of traditional risk factors.12, 13 It is therefore suggested
that apolipoprotein B, as a measure of the number of atherogenic
particles in plasma, may yet provide more information than the
amount of cholesterol transported by these particles.
Insulin and the Risk of IHD
The concept of insulin resistance as a central component of a
potentially atherogenic dyslipidemic state was first introduced in 1988
when it was suggested that a large proportion of individuals resistant
to the action of insulin was also characterized by metabolic
disturbances highly predictive of an increased IHD risk.34 Using
fasting or postglucose insulin levels as crude indices of insulin
resistance, univariate analyses of large cohorts of nondiabetic
populations have shown that hyperinsulinemia in the fasting state or
following a glucose load was associated with an increased risk of
IHD.35-37 Results from multivariate analyses have, however, yielded
discordant conclusions. We11 and others38 have recently reported
that elevated plasma insulin levels measured with an antibody
showing essentially no cross-reactivity with proinsulin were
associated with an increased risk of developing IHD, independent of
other risk factors such as triglyceride, HDL-C, and LDL-C levels.
Nevertheless, whether plasma insulin should or should not be
considered an independent risk factor for the development of IHD
remains a matter of considerable debate. It is well accepted, however,
that elevated plasma insulin concentrations are most frequently
associated with deteriorations in other cardiovascular risk factors.39
Hyperinsulinemia and insulin resistance also appear to have direct
effects on the arterial wall and contribute to a reduced fibrinolytic
potential.40 Plasma insulin levels may therefore provide a crude but
global description of a number of additional metabolic abnormalities
that may, in turn, be associated with an increased risk of IHD, but
that may not be adequately assessed by the traditional triad of lipid
risk factors. It is important to emphasize that results of the present
study apply to nondiabetic men, particularly because patients with
type 2 diabetes mellitus were excluded from the analyses. Although
inclusion of men with type 2 diabetes mellitus in the study sample
essentially had no impact on the results, whether results of the
present study can be applied to other populations such as persons
with type 2 diabetes mellitus, women, or the elderly population will
have to be established more specifically in future studies.
Conclusions
Beyond the mechanisms underlying the atherogenicity of
hyperinsulinemia, hyperapobetalipoproteinemia, and small, dense
LDL, and irrespective of whether these mechanisms share common
paths, results of the present study suggest that the risk of IHD is
increased substantially when these metabolic abnormalities cluster.
The synergistic contribution of the nontraditional cluster of risk factors
to IHD risk and the fact that almost 1 of every 2 IHD cases had these
abnormalities simultaneously reflect the multifactorial etiology of IHD.
It also emphasizes the importance of defining the risk of IHD based
on more than 1 risk factor.
There are a number of critical issues that have to be considered
before any decision can be made toward the measurement of these
nontraditional risk factors on a routine basis. Among others, results of
this prospective case-control study will have to be confirmed through
larger population-based studies, as the relatively low number of IHD
cases allowed only a gross assessment of risk. The relatively large
CIs associated with the estimated risk in some of the subgroups
reflect this phenomenon. Population reference values such as those
used for LDL-C, triglycerides, and HDL-C also will be needed before
critical levels of fasting insulin, apolipoprotein B levels, and LDL
particle size or density at which a person becomes at greater risk for
IHD are identified. Means to achieve effective treatment of the
nontraditional risk factors is also a critical issue that deserves a great
deal of scrutiny before decisions can be made toward use of these
variables in the risk management of IHD. There are data to suggest
that LDL particle size can be modulated by changes in plasma
triglyceride levels.41 Studies have shown that triglyceride-lowering
therapy with fibric acid derivatives can lead to a significant increase
in
LDL particle size.42, 43 There is also a large body of evidence
demonstrating that LDL particle size, apolipoprotein B level, and
insulin resistance and/or hyperinsulinemia can be effectively altered
by diet and exercise-induced weight loss.44, 45 Thus, the ability to
favorably modify the nontraditional risk factors by diet, exercise, and
appropriate pharmacotherapy provides further support for the use of
these risk factors in the management of IHD risk. Finally, the
cost-effectiveness of implementing and using new risk factors as a
basis for screening and treatment in primary and secondary
prevention of IHD should be established. Irrespective of these
important considerations, we hope that these results will help
stimulate research aimed at identifying means that could
substantially improve the early diagnosis and treatment of individuals
at risk for IHD.
Author/Article Information
From the Lipid Research Center, Laval University Hospital Research
Center, Ste-Foy, Québec (Drs Lamarche, Tchernof, Mauriège,
Cantin,
Lupien, and Després); and the Department of Medicine, University
of
Montréal, Montréal, Québec (Dr Dagenais).
Reprints: Jean-Pierre Després, PhD, Lipid Research Center, CHUL
Research Center, 2705 Laurier Blvd, TR-93, Ste-Foy, Québec GIV
4G2, Canada (e-mail: jean-pierre.despres@crchul.ulaval.ca).
This study was supported in part by the Heart and Stroke Foundation
of Canada, the Medical Research Council of Canada, and the Québec
Heart Institute Research Foundation. Dr Lamarche is a research
scholar of the Medical Research Council of Canada and Dr Tchernof
is a recipient of a research fellowship from the Canadian Diabetes
Association.
We are grateful to France Gagnon, MSc, and Louise Fleury, MSc, for
their important contribution in the data collection and to Paul-Marie
Bernard, MSc, biostatistician and professor at the department of
Preventive and Social Medicine, Laval University, Ste-Foy, Québec,
for his helpful input regarding data analysis. The financial contribution
of Fournier Pharma Inc, Montréal, Québec, is also gratefully
acknowledged.
REFERENCES
1.
Castelli WP.
Epidemiology of coronary heart disease: the Framingham Study.
Am J Med.
1984;76:4-12.
MEDLINE
2.
Stamler J, Wentworth D, Neaton JD.
Is the relationship between serum cholesterol and risk of premature
death from coronary heart disease continuous and graded? findings in
356222 primary screenees of the Multiple Risk Factor Intervention
Trial (MRFIT).
JAMA.
1986;256:2823-2828.
MEDLINE
3.
Genest JJ, McNamara JR, Ordovas JM, et al.
Lipoprotein cholesterol, apolipoprotein A-I and B and lipoprotein (a)
abnormality in men with premature coronary heart disease.
J Am Coll Cardiol.
1992;19:792-802.
MEDLINE
4.
Ginsburg GS, Safran C, Pasternak RC.
Frequency of low serum high-density lipoprotein cholesterol levels in
hospitalized patients with desirable total cholesterol levels.
Am J Cardiol.
1991;68:187-192.
MEDLINE
5.
Superko HR.
Beyond LDL cholesterol reduction.
Circulation.
1996;94:2351-2354.
MEDLINE
6.
Austin MA.
Plasma triglyceride and coronary heart disease.
Arterioscler Thromb Vasc Biol.
1991;11:2-14.
7.
Lamarche B, Després JP, Moorjani S, Cantin B, Dagenais GR,
Lupien PJ.
Triglycerides and HDL-cholesterol as risk factors for ischemic heart
disease: results from the Québec Cardiovascular Study.
Atherosclerosis.
1996;119:235-245.
MEDLINE
8.
Manninen V, Tenkanen L, Koshinen P, et al.
Joint effects of serum triglyceride and LDL cholesterol and HDL
cholesterol concentrations on coronary heart disease risk in the
Helsinki Heart Study: implications for treatment.
Circulation.
1992;85:37-45.
MEDLINE
9.
Assmann G, Schulte H.
Relation of high-density lipoprotein cholesterol and triglycerides to
incidence of atherosclerotic coronary artery disease: the PROCAM
experience.
Am J Cardiol.
1992;70:733-737.
MEDLINE
10.
Assmann G, Schulte H.
Identification of individuals at high risk for myocardial infarction.
Atherosclerosis.
1994;110(suppl):S11-S21.
MEDLINE
11.
Després JP, Lamarche B, Mauriège P, et al.
Hyperinsulinemia as an independent risk factor for ischemic heart
disease.
N Engl J Med.
1996;334:952-957.
MEDLINE
12.
Lamarche B, Moorjani S, Lupien PJ, et al.
Apolipoprotein A-I and B levels and the risk of ischemic heart disease
during a five-year follow-up of men in the Québec Cardiovascular
Study.
Circulation.
1996;94:273-278.
MEDLINE
13.
Lamarche B, Després JP, Moorjani S, Cantin B, Dagenais GR,
Lupien PJ.
Prevalence of dyslipidemic phenotypes in ischemic heart disease:
prospective results from the Québec Cardiovascular Study.
Am J Cardiol.
1995;75:1189-1195.
MEDLINE
14.
Lamarche B, Tchernof A, Dagenais GR, Cantin B, Lupien PJ,
Després JP.
Small, dense LDL particles and the risk of ischemic heart disease:
prospective results from the Québec Cardiovascular Study.
Circulation.
1997;95:69-75.
MEDLINE
15.
Lamarche B, Moorjani S, Cantin B, Dagenais GR, Lupien PJ,
Després JP.
Associations of HDL2 and HDL3 subfractions with ischemic heart
disease in men: prospective results from the Québec Cardiovascular
Study.
Arterioscler Thromb Vasc Biol.
1997;17:1098-1105.
MEDLINE
16.
Dagenais GR, Robitaille NM, Lupien PJ, et al.
First coronary heart disease event rates in relation to major risk
factors: Québec Cardiovascular Study.
Can J Cardiol.
1990;6:274-280.
MEDLINE
17.
Moorjani S, Dupont A, Labrie F, et al.
Increase in plasma high density lipoprotein concentration following
complete androgen blockage in men with prostatic carcinoma.
Metabolism.
1987;36:244-250.
MEDLINE
18.
Albers JJ, Warnick GR, Wiebe D, et al.
Multi-laboratory comparison of three heparin-MnCl2 precipitation
procedures for estimating cholesterol in high-density lipoproteins.
Clin Chem.
1978;24:323-338.
19.
Friedewald WT, Levy RI, Fredrickson DS.
Estimation of the concentration of low density lipoprotein cholesterol
in plasma, without use of the preparative ultracentrifuge.
Clin Chem.
1972;18:499-502.
MEDLINE
20.
Laurell CB.
Electroimmunoassay.
Scand J Clin Lab Med.
1972;124:23-27.
21.
Krauss RM, Burke DJ.
Identification of multiple subclasses of plasma low density
lipoproteins in normal humans.
J Lipid Res.
1982;23:97-104.
MEDLINE
22.
McNamara JR, Campos H, Ordovas JM, Peterson J, Wilson PWF,
Schaefer EJ.
Effect of gender, age, and lipid status on low density lipoprotein
subfraction distribution: results from the Framingham Offspring Study.
Arterioscler Thromb Vasc Biol.
1987;7:483-490.
23.
Tchernof A, Lamarche B, Nadeau A, et al.
The dense LDL phenotype: association with plasma lipoprotein levels,
visceral obesity and hyperinsulinemia in men.
Diabetes Care.
1996;19:629-637.
MEDLINE
24.
Austin MA, Breslow JL, Hennekens CH, Buring JE, Willet WC,
Krauss RM.
Low density lipoprotein subclass patterns and risk of myocardial
infarction.
JAMA.
1988;260:1917-1921.
MEDLINE
25.
Campos H, Genest JJ, Blijlevens E, et al.
Low density lipoprotein particle size and coronary artery disease.
Arterioscler Thromb Vasc Biol.
1992;12:187-195.
26.
Coresh J, Kwiterovich PO, Smith HH, Bachorik PS.
Association of plasma triglyceride concentration and LDL particle
diameter, density, and chemical composition with premature coronary
artery disease in men and women.
J Lipid Res.
1993;34:1687-1697.
MEDLINE
27.
Gardner CD, Fortmann SP, Krauss RM.
Association of small low-density lipoprotein particles with the
incidence of coronary artery disease in men and women.
JAMA.
1996;276:875-881.
MEDLINE
28.
Stampfer MJ, Krauss RM, Ma J, et al.
A prospective study of triglyceride level, low-density lipoprotein
particle diameter, and risk of myocardial infarction.
JAMA.
1996;276:882-888.
MEDLINE
29.
de Graaf J, Hak Lemmers HL, Hectors MP, Demacker PNM,
Hendriks JC, Stalenhoef AF.
Enhanced susceptibility to in vitro oxidation of the dense low density
lipoprotein subfraction in healthy subjects.
Arterioscler Thromb Vasc Biol.
1991;11:298-306.
30.
Nigon F, Lesnik P, Rouis M, Chapman MJ.
Discrete subspecies of human low density lipoproteins are
heterogeneous in their interaction with the cellular LDL receptor.
J Lipid Res.
1991;32:1741-1753.
MEDLINE
31.
Hodis HN, Mack WJ, Azen SP, et al.
Triglyceride- and cholesterol-rich lipoproteins have a differential effect
on mild/moderate and severe lesion progression as assessed by
quantitative coronary angiography in a controlled trial of lovastatin.
Circulation.
1994;90:42-49.
MEDLINE
32.
Davignon J, Cohn JS.
Triglycerides: a risk factor for coronary heart disease.
Atherosclerosis.
1996;124(suppl):S57-S64.
MEDLINE
33.
Sniderman AD, Cianflone K.
Measurement of apoproteins: time to improve the diagnosis and
treatment of the atherogenic dyslipoproteinemias.
Clin Chem.
1996;42:489-491.
MEDLINE
34.
Reaven GM.
Role of insulin resistance in human disease.
Diabetes.
1988;37:1495-1507.
35.
Yarnell JWG, Sweetnam PM, Marks V, Teale JD, Bolton CH.
Insulin in ischaemic heart disease: are associations explained by
triglyceride concentrations? the Caerphilly Prospective Study.
Br Heart J.
1994;71:293-296.
MEDLINE
36.
Pyörälä K.
Relationship of glucose tolerance and plasma insulin to the incidence
of coronary heart disease: results from two population studies in
Finland.
Diabetes Care.
1979;2:131-141.
MEDLINE
37.
Eschwège E, Richard JL, Thibult N, et al.
Coronary heart disease mortality in relation with diabetes, blood
glucose and plasma insulin levels: the Paris prospective study, ten
years later.
Horm Metab Res.
1985;15:41-46.
38.
Perry IJ, Wannamethee SG, Whincup PH, et al.
Serum insulin and incident coronary heart disease in middle-aged
British men.
Am J Epidemiol.
1996;144:224-234.
MEDLINE
39.
Haffner SM, Mykkänen L, Stern MP, Valdez RA, Heisserman JA,
Bowsher RR.
Relationship of proinsulin and insulin to cardiovascular risk factors in
nondiabetic subjects.
Diabetes.
1993;42:1297-1302.
MEDLINE
40.
Juhan-Vague I, Alessi MC, Vague P.
Increased plasma plasminogen activator inhibitor 1 levels: a possible
link between insulin resistance and atherothrombosis.
Diabetologia.
1991;34:457-462.
MEDLINE
41.
McNamara JR, Jenner JL, Li Z, Wilson PW, Schaefer EJ.
Change in LDL particle size is associated with change in plasma
triglyceride concentration.
Arterioscler Thromb Vasc Biol.
1992;12:1284-1290.
42.
Yuan JN, Tsai MY, Hunninghake DB.
Changes in composition and distribution of LDL subspecies in
hypertriglyceridemic and hypercholesterolemic patients during
gemfibrozil therapy.
Atherosclerosis.
1994;110:1-11.
MEDLINE
43.
Guerin M, Bruckert E, Dolphin PJ, Turpin G, Chapman MJ.
Fenofibrate reduces plasma cholesteryl ester transfer from HDL to
VLDL and normalizes the atherogenic, dense LDL profile in combined
hyperlipidemia.
Arterioscler Thromb Vasc Biol.
1996;16:763-772.
MEDLINE
44.
Williams PT, Krauss RM, Vranizan KM, Wood PD.
Changes in lipoprotein subfractions during diet-induced and
exercise-induced weight loss in moderately overweight men.
Circulation.
1990;81:1293-1304.
MEDLINE
45.
Després JP, Lamarche B.
Effects of diet and physical activity on adiposity and body fat
distribution: implications for the prevention of cardiovascular disease.
Nutr Res Rev.
1993;6:137-159.
GEN
The gene length is 43 kb. Exons: 29; introns: 28. Two exons are unusually
long: 1.9 kb (the 29th)
and 7.6 kb (the 26th); the length of the remaining exons vary within the
limits of 150-250 bp. The
mature mRNA, 14.1 kb in length, codes for the protein comprising 4563 amino
acid residues."
FUN
[1] Circulatory APOB is a ligand for the receptor-mediated transition of
very low density
lipoproteins (VLDL particles) into cells and plays the central role in
the transport and metabolism
of serum cholesterol.
[2] In addition, APOB, along with glycoprotein Lp(a) (GEM:06q27/APOLPA),
participates in
formation of the complex of serum lipoprotein(a), which plays an important
role in atherogenesis
and pathogenesis of coronary disease."
MOP
APOB has two forms: (I) apoB100 (500 kD; 14.1 kb mRNA) is synthesized in
liver and participates
in the packing of VLDL particles, (II) apoB48 (210 kD; 7.5 kb mRNA) is
synthesized in intestine
and participates in formation of chylomicrons (Chen-1987). ApoB-48 represents
the amino-terminal
47% of apoB-100 and that the carboxyl terminus of apoB-48 is in the vicinity
of residue 2151 of
apoB-100 (Innerarity-1987). ApoB-48 contains 2,152 residues compared to
4,535 residues in
apoB-100 (Higuchi-1988). Both forms are alternative splicing products of
the single gene
(Cladaras-1986)."
Apolipoprotein
(apo) E is a 34-kDa protein consisting of 299 amino acids. It is a protein
constituent of chylomicrons, very low density lipoproteins and HDL and
VLDL remnants (Mahley 1988). On these particles, apo E serves as a ligand
for uptake by lipoprotein receptors (Davignon et al. 1988, Mahley 1988,Mahley
et al. 1990). Apo E is polymorphic with three common alleles: E2, E3 and
E4
(Zannis
et al. 1982), which are associated with variations in the blood lipid concentrations.
The phenotype E2/2 is associated with type III hyperlipidemia, and E4 is
associated with elevated serum total and LDL cholesterol concentrations
compared to E2 and E3 (Ehnholm et al. 1986, Utermann 1987, Davignon et
al. 1988). The serum concentrations of apo E are higher in individuals
with E 3/3 than in individuals with E4, and highest in individuals with
E2 (Berglund et al. 1993, Luc et al. 1994).
Apo
E polymorphism modifies plasma lipids, at least in Caucasians, partly by
affecting the efficiency of cholesterol absorption, so that individuals
with E2 absorb less cholesterol than individuals with E4 (Kesäniemi
et al. 1987), and individuals with the E4 allele respond better to changes
in dietary cholesterol and saturated fatty acids than those without the
E4 allele (Lehtimäki et al. 1995). The apo E polymorphism also modifies
the metabolism of LDL. Individuals with the apo E phenotype 2/2 catabolise
LDL faster than others, and normolipemic apo E 4 homozygotes catabolise
LDL at a slower rate than apo E 3 homozygotes (Demant et al. 1991).
The
E4 phenotype has been associated with an increased risk of CAD either directly
(Kuusi et al. 1989) or via elevated atherogenic lipoprotein levels (Stuyt
et al. 1991). The apo E phenotype distribution among CAD patients and myocardial
infarction survivors is controversial. Some studies propose a higher frequency
of apo E 4 in CAD patients (Nieminen et al. 1992, Wang et al. 1995), or
myocardial infarction (AMI) survivors (Cumming and Robertson 1984), whereas
others fail to detect any difference (Stuyt et al. 1991, Utermann et al.
1984).