In dieser Diplomarbeit wurde gezeigt, daß die elektrochemische Reduktion von Chrom(III)-oxychlorid in nichtwässrigen Elektrolytlösungen möglich ist.
Das Ausgangsmaterial wurde durch Gasphasentransport hergestellt.
Es gelang, das Verfahren soweit zu optimieren, daß auf eine nachträgliche, wässrige Aufarbeitung verzichtet werden konnte.
Diese Voraussetzung war für die elektrochemischen Untersuchungen wichtig.
Bei Syntheseversuchen, die von CrCl3 und TiO2 ausgingen, wurde eine Reihe bisher nicht bekannter, flüchtiger, bis ca. -40°C stabiler Produkte gefunden; die Bildung von CrOCl aber konnte trotz Variation verschiedener Parameter nicht erreicht werden.
Zur Auswertung der erhaltenen Massenspektren der Produkte wurde ein Fortran-Programm geschrieben, das die Simulation von Isotopenmustern unterschiedlichster Spezies erlaubt.
Damit konnten zugeordnet werden:
HCl Cl2 TiCl4 HCrCl4 HCrCl6 HCrCl8
und deren Abbauprodukte.
Die endgültige Synthese erfolgte in Anlehnung an das Verfahren von H.Schäfer und F.Warthenpfuhl(1), mußte jedoch in einigen experimentellen Details geändert werden.
Das so erhaltene CrOCl wurde der elektrochemischen Reduktion unterworfen, wobei es gelang, eine Reihe nichtstöchiometrischer Intercalationsverbindungen vom Typ
M+x (DMSO)y Cr2+x Cr3+1-x (OCl)-
mit M = Li, Na und K darzustellen, die anhand von Debye-Scherrer-Aufnahmen röntgenographisch charakterisiert wurden.
Dabei zeigte sich in allen bisher untersuchten Fällen aus den Gitteraufweitungen der reduzierten Verbindungen, daß die solvatisierten Kationen eingelagert wurden.
Die mangelhafte Integralität der Röntgenreflexe wies auf stark gestörte Produkte hin.
Die galvanostatische Reduktion zeigte in der Regel sehr gut ausgebildete Potentialstufen.
Der Reduktionsgrad lag meist bei 15%.
Das Bild der Potentialstufen war abhängig von der gewählten Stromdichte, die zwischen 20 und 120 µA variiert wurde.
Die Ladungsverluste bei Umkehr der Reaktion waren erheblich; das Gitter wurde schon unter schwach oxidierenden Bedingungen (+0.2V vs. SCE) zerstört.
Verfolgt man die einschlägige Literatur und ausgelegte Patente auf dem Gebiet der Festkörperchemie, so beobachtet man, daß Publikationen über Intercalation in Schichtverbindungen einen immer größeren Raum einnehmen.
Lange Zeit waren als einlagerungsfähige Verbindungen nur bestimmte Tonmineralien (Kaolin, glimmerartige Schichtsilikate) bekannt.
Etwa ab 1945 kam hierzu in Form von Graphitverbindungen eine neue Gruppe von Intercalationskomplexen mit elektronenleitendem Wirtsgitter.
Im Unterschied zu den Komplexen der Tone erfolgt bei diesen leitenden Wirtsgittern während der Einlagerung zwischen Intercalat und Wirtsgitter häufig ein Redoxprozeß, dessen essentielle Bedeutung für die Intercalation erst in neuerer Zeit Untersuchungsgegenstand wurde.
Der Elektronenübergang kann durch das Intercalat selbst, durch Zugabe eines chemischen Reagenzes oder durch Anlegen eines Potentials an das Wirtsgitter auch erzwungen werden.
Hierzu kamen später Verbindungen, die schlechte Elektronenleiter waren, aber bei beginnender Oxidation oder Reduktion ihre Leitfähigkeit durch Elektronenleitung (n- oder p-Leitung) um Größenordnungen erhöhen.
Die mit einem Elektronenübergang verbundene Intercalation bot die Möglichkeit, Intercalationsverbindungen als elektroaktive Komponenten in Batterien einzusetzen.
Neuere Untersuchungen zeigten, daß sich in vielen längst bekannten, kommerziellen Batterien und Akkumulatoren Intercalationsprozesse abspielen:
| |
MnO2 + H3O+ |
--> |
(H3O+)x MnO2 |
|
(Leclanché) |
| |
PbO2 + H3O+ |
--> |
(H3O+)x PbO2 |
|
(Bleiakkumulator) |
Auch die Bronzen des Vanadin, Wolfram und Molybdän lassen sich als Einlagerungsverbindungen beschreiben und haben als solche bereits praktische Bedeutung erlangt:
In Display-Anzeigen wird die Farbändeurng von farblos nach blau in folgender Reaktion verwandt:
| |
x H+ + e- + WO3 |
--> |
HxWO3 |
|
(2,
3,
4) |
Eine Unterteilung der Intercalationskomplexe ist leicht durch die elektronische Wechselwirkung zwischen Wirtsgitter und Intercalat möglich.
Speziell durch die elektrochemische Präparation ist es möglich, in gewissen Grenzen Intercalat und Stöchiometrie (Nichtstöchiometrie) zu variieren.
Durch mehr oder weniger starke Donor-Akzeptor-Wechselwirkungen zwischen Wirtsgitter und Intercalat treten Änderungen der elektronischen Eigenschaften auf, die die Bandenstruktur beeinflussen.
Durch die Einlagerung wird das Wirtsgitter aufgeweitet; dies wiederum verändert das Elektronen- und Phononenspektrum.
Hier liegen Ansätze zu theoretischen Arbeiten.
Es sind aber auch experimentelle Arbeiten zu erwähnen:
z.B. die Eignung dieser Schichtverbindungen als Supraleiter:
relativ bald stellte man fest, daß die Intercalation die Sprungtemperaturen mancher Wirtsgitter bemerkenswert verändern konnte.
In dieser Hinsicht untersuchte Modellsubstanzen sind 2H-TaS2 und NbS2. Auch die Chevrel-Phasen PbxMo6S8-y mit maximalen Sprungtemperaturen um 15K(5) gehören zu den Intercalationsverbindungen.
Schließlich haben sich gewisse Einlagerungsverbindungen wegen ihrer großen inneren Oberfläche in der heterogenen Katalyse bewährt:
Durch die Anwesenheit des Wirtsgitters kann die chemische Aktivität der eingelagerten Molekel so verändet werden, daß sie zu sonst schwer erreichbaren Reaktionen fähig ist.
Voraussetzung ist hierbei eine schnelle reversible Einlagerung und große Beweglichkeit auch größerer Teilchen unter Ausnützung der gesamten inneren Oberfläche.
Als Beispiel mögen Intercalationsverbindungen des Graphit dienen, denen unter bestimmten Voraussetzungen die Rolle eines Katalysators z.B. in der Ammoniaksynthese(6), der Darstellung von Kohlenwasserstoffen aus CO und H2(7) und vielen anderen problematischen Syntheseschritten zugesprochen wird.
Katalytische Anwendungen der Intercalationskomplexe sind Gegenstand vieler neuerer Artikel(8).
In diesem breiten Feld der chemischen Möglichkeiten stellt das in dieser Arbeit untersuchte Chrom(III)-oxychlorid einen Grenzfall dar.
CrOCl muß wegen seiner Struktur(9) zu den Schichtverbindungen gerechnet werden.
J.Choy und A.Weiss(10) stellten fest, daß eine chemische Intercalation in CrOCl nur nach einer vorangegangenen Reduktion der Kristalloberfläche möglich ist.
Da andererseits von den isotypen Verbindungen FeOCl, TiOCl und VOCl Einlagerungskomplexe mit Ammoniak, Aminen und Alkoholen bekannt und ohne Schwierigkeiten darzustellen sind, scheint bei CrOCl eine Grenze für Intercalationsreaktionen erreicht zu sein.
Die Aufgabenstellung in dieser Arbeit bestand darin, dieses Verhalten mit elektrochemischen Methoden zu charakterisieren.
Gleichzeitig sollte mit diesem Verfahren gezeigt werden, ob die Einlagerung in CrOCl umkehrbar (d.h., daß bei Potentialumkehr die eingelagerte Substanzmenge quantitativ ausgelagert wird) oder reversibel (d.h. im thermodynamischen Gleichgewicht ohne kinetische Hemmung) verläuft.
In diesem Falle könnte die Eignung von CrOCl als Speicher- und Transportelement in Primär- und Sekundärzellen geprüft werden.
Da Batterien, je nach Anwendungsfeld, einen Kompromiß aus den Größen Energiedichte, Leistungsdichte, Lagerfähigkeit, Herstellungskosten und - im Falle von Akkumulatoren - Anzahl der möglichen Lade/Entladezyklen darstellen, erlauben vergleichende Untersuchungen mit bekannten Systemen eine unmittelbare Möglichkeit, einen eventuellen Anwendungsbereich zu klassifizieren.
Eine Entwicklung neuer Zellen kann an drei Stellen ansetzen:
Jedes spannungsliefernde Element besteht aus der elektropositiven Anode ( = negative Batterieelektrode), der elektronegativen Kathode ( = positive Batterieelektrode) und einem Elektrolyten in flüssiger, verfestigter oder fester Form für den Ladungstransport.
Die Chemie der Schichtverbindungen leistet zu allen Komponenten ihren Beitrag.
Gewöhnlich besteht die Anode bei Primärzellen zur Erhöhung der Zellspannung aus elektropositiven Metallen wie Lithium oder Natrium.
In Sekundärelementen treten mit diesen Anoden Schwierigkeiten auf, da sich beim Ladevorgang das Alkalimetall leicht dendritisch abscheidet, wodurch wegen der geringeren mechanischen Festigkeit der Dendriten die Lebensdauer und erreichbare Zyklenzahl des Elements erheblich eingeschränkt wird.
Diese Schwierigkeiten können (neben einem Ausweichen auf weniger elektropositive Anoden) durch Einbau der Alkaliionen in ein Wirtsgitter umgangen werden.
Die Legierungen LixAl mit x = 1...2 sind hier vielversprechende Verbindungen.
An Kathoden sind Einlagerungen, wie bereits erwähnt, keineswegs ungewöhnlich und oft charakteristisch für das verwendete Element (die schnelle Protoneneinlagerung in PbO2 ist für die hohe Leistungsdichte des Bleiakkumulatores von großer Bedeutung).
Zur Klassifizierung der Schichtverbindungen bezüglich Eignung als Anoden bzw. Kathoden sind einige Kriterien bedeutsam(11):
- leichte Diffusion des Intercalats in das Gitter und große Beweglichkeit im Gitter bei niedrigen Temperaturen (Leistungsdichte, Reversibilität)
- große Phasenbreite des Intercalationskomplexes mit geringer Änderung der Bildungsenthalpie (Leistungsgewicht, Potentialkonstanz)
- geringe strukturelle Veränderungen während der Intercalation (mechanische Stabilität, möglichst keine Solvateinlagerung, Reversibilität(12))
- reversible Intercalation (Eignung als Sekundärelektrode, Lebensdauer, Zyklenzahl)
- elektronische Leitfähigkeit (Leistungsdichte)
- Unlöslichkeit des Intercalationskomplexes im Elektrolyten und chemische Beständigkeit in einem weiten Potentialbereich (Lebensdauer)
- geringer Einfluß der Temperatur auf alle obigen Kriterien (Konstanz der physikalischen Eigenschaften)
Für Anoden läßt sich zusätzlich fordern:
- möglichst geringe Änderung des Anodenpotentials durch die Intercalation (Energiedichte);
für Kathoden:
- große freie Bildungsenthalpie der Intercalationsverbindungen (Energiedichte)
Die für Kathodenmaterialien angegebenen Kriterien werden von Schichtverbindungen teilweise relativ gut erfüllt und das System TiS2/LixTiS2 war auch schon für kommerzielle Anwendung im Gespräch(12, 13, 14, 15).
Schlechte Lagerfähigkeit im entladenen Zustand beendeten schnell die Hoffnungen an dieses Kathodenmaterial.
Aber auch die meisten der bisher untersuchten Dichalcogenide zeigen elektrochemische Aktivität in Lithiumzellen(16, 17, 18, 19), sodaß, besonders für spezielle Anforderungen wie z.B. ein bestimmter Strom- oder Spannungsverlauf während der Entladung, noch ein weites Feld der kommerziellen Anwendung offen leibt.
Für den Einsatz von CrOCl als Batterieelektrode wird wegen des Redoxpotentials Cr3+/Cr2+ (E1/2 = -0,5...-1,2V vs. SCE(20), das im CrOCl nicht sehr verschieden sein sollte, nur eine Verwendung als Kathode zur Diskussion stehen.
Sie gewährleisten den Ionentransport zwischen den Elektroden.
Dieser Vorgang soll leicht und im Idealfall ausschließlich für eine Ionensorte funktionieren;
eine Elektronenleitung soll aber vollständig ausgeschlossen werden.
Ein kommerziell genutztes und sehr gut untersuchtes System mit Schichtstruktur ist Natrium-β-Alumina(21).
Die Verwendung von CrOCl in diesem Sinne verlangt einen schnellen Transport der Alkaliionen in den Schichten, das bedeutet eine sehr hohe Beweglichkeit der eingelagerten Kationen.
Bei den isotypen Strukturen FeOCl(22) und VOCl(23) ist bekannt, daß diese Voraussetzungen nur mangelhaft erfüllt sind.
Bei der Suche nach potentiellen Batterieelektroden führte 1976 M. S. Whittingham an einigen neuen Klassen von Schichtverbindungen, zu denen auch FeOCl gehörte, elektrochemische Voruntersuchungen durch(24, 16, 25).
Schnell folgten Arbeiten über VOCl, VOBr, CrOCl und CrOBr(26, 25);
das chemische und kristallographische Interesse an diesen Verbindungen trat jedoch in den Hintergrund.
Durch die vielversprechenden Ergebnisse bei den Dichalcogeniden (Patenterteilungen loc. cit. 18) genügten zur Untersuchung auf Eignung als Elektrode Cyclovoltagramme ohne weitere Charakterisierung der erhaltenen Verbindungen, um eventuelle patentrechtliche Ansprüche zu sichern.
Die Autoren führten diese Reduktionen in Lithiumzellen mit LiClO4 in Acetonitril als Elektrolytlösung durch.
Elektrochemische Untersuchungen mit Charakterisierung der erhaltenen Komplexe wurden durchgeführt von
R. Schöllhorn / H. Meyer an TaS2(17)
J. Dahn / R. R. Haering an TiS2(27)
H. Meyer / A. Weiss / J. O. Besenhard an FeOCl(22) und
J.. P. Venién et al. an VoCl(23).
Für die Darstellung von CrOCl sind in der Literatur eine Reihe von Verfahren beschrieben
(1, 10, 28, 29, 30, 31):
| a) |
Cr2O3 |
|
+ |
|
CrCl3 |
(1) |
| b) |
TiO2 |
|
+ |
|
CrCl3 |
(1, 28) |
| c) |
H2O |
|
+ |
|
CrCl3 |
(1) |
| d) |
CrO3 |
+ |
Cr |
+ |
CrCl3 |
(1) |
| e) |
Bi2O3 |
|
+ |
|
CrCl3 |
(1) |
| f) |
SiO2 |
|
+ |
|
CrCl3 |
(1) |
| g) |
Cr2O3 |
|
+ |
|
SiCl4 |
(1) |
| h) |
O2 |
|
+ |
|
CrCl3 |
(31) |
Zur Entfernung von überschüssigem CrCl3 schlägt Schäfer die Behandlung der Reaktionsprodukte mit zehnprozentiger wässriger CrCl2-Lösung vor, wodurch CrCl3 in Lösung geht, CrOCl wegen seiner Reaktionsträgheit ohne sichtbare Schädigung verbleibt.
Eine partielle Reduktion oder Hydrolyse von CrOCl kann aber nicht ausgeschlossen werden.
Eine derartige Veränderung von CrOCl hätte zur Folge, daß bei den elektrochemischen Versuchen zusätzliche Potentiale auftreten, die die Interpretation der Cyclovoltagramme und Galvanostatiken sehr erschweren würden.
Zur Optimierung erschienen die Synthesen a) und b) als zweckmäßig, da einer Verunreinigung von CrOCl durch Nebenprodukte mit geringem Aufwand begegnet werden konnte und die Ausbeuten relativ hoch sein sollten.
Das zur Synthese eingesetzte Chrom(III)-oxychlorid (Merck 27094 wasserfrei) wurde durch Transport im trockenen und weitgehend sauerstofffreien Chlorstrom (ca. 1ltr./min.) bei 920°C durch 'Sublimation' gereinigt.
Ein Transport bei höherer Temperatur brachte nicht die gewünschte Trennung von leichter flüchtigen Komponenten und führte zu einem fein verteilten Pulver.
Durch Aufwachsen der Kristalle konnte bei zu schwachem Chlorstrom die gesamte Rohröffnung versperrt werden.
Einne sparsame und bequeme Methode bestand darin, den Chlorstrom durch Hin- und Herkondensieren desChlors zwischen zwei Fallen zu erzeugen.
Die Trocknung des Chlors erfolgte an P4O10/Vermiculit.
Der Sauerstoff wurde durch Ausfrieren des Chlors bei ca. -60°C und Spülung mit getrocknetemStickstoff weitgehend entfernt (nicht erfaßt wurde der im flüssigen Chlor gelöste Sauerstoff!).
Als TiO2 wurde eine Anatas-Modifikation (Degussa P25III) eingesetzt, die bei 200°C imHochvakuum (10-5Torr) von anhaftenden Feuchtigkeitsresten weitgehend befreit wurde.
Die eingewogenen Ausgangskomponenten wurden unter Argon in eine Kunststoffolie eingeschweißt, in einer isostatischen Presse (30to/cm²) gepreßt und in das Reaktionsgefäß aus Quarz überführt.
Alle diese Manipulationen wurden in einer Argonkammer (bei Dr. Evers) ausgeführt, um eine Kontamination mit Wasser soweit wie möglich zu verhindern.
Dieser aufwendige Feuchtigkeitsausschluß war notwendig, da sonst während der Aufheizphase die Ampullen wegen gebildetem HCl zur Explosion neigen und teilweise hydrolyseempfindliche Produkte beobachtet wurden (siehe unten).
Die massenspektroskopischen Untersuchungen zeigten jedoch, daß trotz dieser Maßnahmen noch Chlorwasserstoff entstand;
das hierzu notwendige Wasser wurde sehr wahrscheinlich durch TiO2 eingeschleppt;
die spezifischeOberfläche des verwendeten TiO2 betrug ca. 39 m²/g(32).
Die Daten der Syntheseversuche und die experimentellen Details sind in Tabelle 3.1 zusammengefaßt.
Bei den Ansätzen konnten je nach Bedingungen bis zu sechs Produkte unterschieden werden:
Die Ergebnisse im einzelnen (siehe auch Tab 3.1):
| Versuch 1.1 |
Beim Aufheizen auf 800°C schlug sich in der Falle neben großen Mengen TiCl4 eine wesentlich kleinere Menge einer stark lindgrün gefärbten, flüchtigen Substanz nieder.
Diese war instabil und zersetzte sich teilweise bei der Sublimation im Vakuum (p = 10-5Torr, T = -35°C).
Bei diesem Zerfall entstand ein farbloses, extrem hygroskopisches Produkt.
Die energiedispersive Analyse im Rasterelektronenmikroskop zeigte die Anwesenheit von Chrom und Titan im ungefähren Verhältnis 1:4...9 und einen hohen Chlorgehalt.
Eine Chlorbestimmung (mit AgNO3, Endpunkt mit CrO4-) ergab (61,7+/-1,5) Gew.% CL-.
Die Angabe einer Stöchiometrie wäre wegen der fehlerbehafteten Analyseergebnisse noch verfrüht.
Die Guinier-Aufnahmen des grünen, nicht zersetzlichen Produkts aus dem Reaktionsrohr zeigte, daß auf diese Weise neben geringen Spuren (ca. 3%, unter der Stereolupe gefunden) CrOCl nur Cr2O3 dargestellt wurde.
Am Ofenende befand sich im Reaktionsrohr etwas sublimiertes CrCl3.
|
| Versuch 1.2 |
Zur Auftrennung der flüchtigen Reaktionsprodukte wurden zwei Fallen eingesetzt.
Neben etwas Chlorund TiCl4 konnte die flüchtige grüne Verbindung in der Falle mit LN2-Kühlung gefunden werden.
Versuche, am Massenspektrometer Aufschlüsse über diese Substanz zu bekommen, scheiterten an der Störung durch den zu großen Überschuß an TiCl4.
Die Analyse der wie bei Versuch 1.1 erhaltenen hygroskopischen Verbindung ergab (innerhalb der Fehlergrenzen) diesselben Werte.
|
| Versuch 1.3 |
Da offensichtlich durch die angeschlossene Hochvakuumpumpe der Gleichgewichtsdruck von CrCl3 in Versuch 1.1 und 1.2 so stark erniedrigt wurde, daß eine Zersetzung von CrOCl eingesetzt haben könnte (Christensen erreichte während der Reaktion mit laufender Quecksilberdiffusionspumpe nicht einmal ein Torr!) oder ein Reaktionspartner der Reaktion entzogen wurde, haben wir die Apparatur vor dem H
eizen nach Erreichen eines Vakuums von p = 10-4 Torr von der Hochvakuumpumpe getrennt.
Das Reaktionsrohr blieb allerdings mit den Kühlfallen verbunden.
Eine Überprüfung des Produkts nach einer Reaktionszeit von zwei Stunden bei 900°C ergab nur Cr2O3.
Am Ofenende befand sich wieder etwas CrCl3;
die lindgrüne Verbindung konnte in den Fallen aber nicht gefunden werden.
|
| Versuch 1.4 |
Nachdem eventuell auch die Falle im vorhergehenden Versuch den Partialdruck einer wichtigen Reaktionskomponente soweit herabgesetzt haben könnte, daß die Reaktion zum gewünschten Produkt gehemmt wurde oder das Produkt sich wieder zersetzte, wurde in diesem Versuch auch auf eine Falle verzichtet.
Die Ampulle wurde nach dem Evakuieren (10-5 Torr) abgeschmolzen und so in den Ofen gelegt, daß ein Ende ca. 1cm herausragte.
Aber auch dieser Versuch führte nur zu Cr2O3.
Beim Herausnehmen der Ampulle zeigte sich die in Abb 3.1 dargestellte Produktverteilung.
|
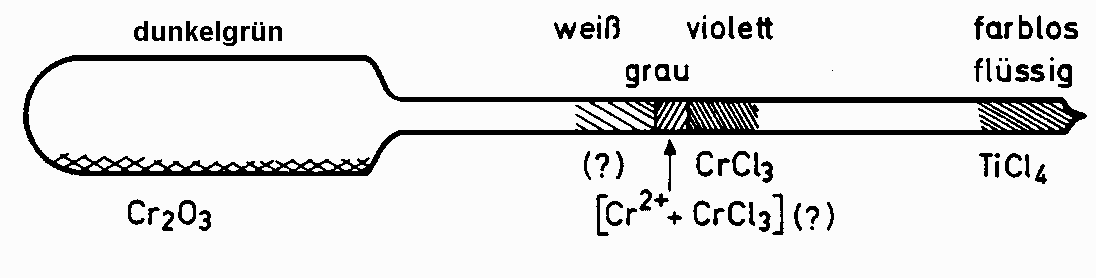
Abb 3.1: Produktverteilung aus Versuch 1.4
|
| Versuch 1.5 |
Da, wie die Versuche 1.1 bis 1.4 zeigten, außer den beschriebenen Produkten Cr2O3, CrCl3, TiCl4, geringe Mengen CrOCl, aber unter bestimmten Voraussetzungen eine Reihe unbekannter, flüchtiger Verbindungen entstanden, wurde der Versuch 1.5 unter denselben Bedingungen wie 1.1 am Massenspektrometer durchgeführt.
Damit sollten Verluste der zersetzlichen Produkte vermieden werden.
Die erhaltenen Massenspektrogramme zeigten - in Abhängigkeit von der Temperatur - den Fortgang der Reaktion (vergl. Abb 3.2):
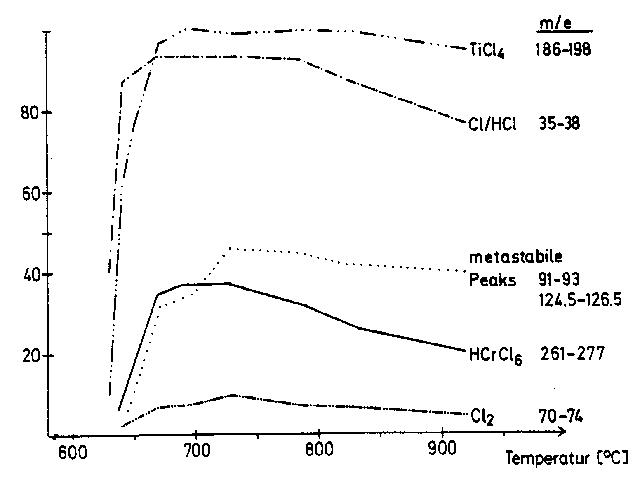 Abb 3.2
Abb 3.2: Gehalt signifikanter Bruchstücke im Spektrogramm Auftretendes TiCl4 ab ca. 620°C weißt auf den Beginn der Reaktion hin.
Gleichzeitig treten die Signale von Cl, HCl und CL2 auf.
Ab ca. 650°C treten in der Region m/e = 261...277 Signale auf, die durch ihr Muster einem Fragment 'HCrCl6' zugeordnet werden können.
Eine ähnliche Gruppe tritt bei m/e = 331...351 auf (möglicherweise 'HCrCl8').
Zwei metastabile (weil stark verbreitert) Peaks um m/e = 91...93 und m/e = 124,5...126,5 konnten wir bisher nicht zuordnen.
Ab ca. 670°C tauchen in vielen Bereichen des Spektrums relativ starke Signale auf, deren Zuordnung uns bisher nicht gelungen ist;
aus dem Muster dieser Signale läßt sich auf einen hohen Gehalt an Chlor und etwas Chrom schließen.
Bis zum Ende des Versuchs bei ca. 920°C können die beschriebenen Signale beobachtet werden.
Ein Teil der unbekannten Verbindungen läßt sich als Fragmente einer Titan-Chrom-Verbindung interpretieren, aber eine sichere Aussage ist wegen der geringen Intensität dieser Signale noch nicht möglich.
Dieses Ergebnis würde sich aber mit den Analysen aus Versuch 1.1 decken.
|
Flüchtige gemischte Titan-Chrom-Halogenide sind bisher in der Literatur nicht beschrieben, ebenfalls nicht bekannt sind Bruchstücke 'HCrCl6' und 'HCrCl8'.
Da aber gleichzeitig Cl2 im Massensprektrum beobachtet wurde, sind Redoxprozesse während dieser Reaktion nicht ausgeschlossen.
Die in Versuch 1.1 gefundene lindgrüne Substanz konnte wegen ihrer Zersetzlichkeit mit keinem Signal im Massenspektrogramm zweifelsfrei korreliert werden.
Aus den Ergebnissen der Versuche 2.1 bis 2.7 (siehe 3.2) ist es denkbar, daß intermediär entstandenes CrOCl wegen des sehr niedrigen Partialdrucks von CrCl3 im System (die kühlste Stelle der Apparatur hatte höchstens eine Temperatur von ca. 200°C; Versuch 1.4) in Umkehrung der unter 3.2 beschriebenen Reaktion in CrCl3 und Cr2O3 zerfallen ist.
Zur Isolierung der Nebenprodukte und deren Untersuchung wäre viel Zeit zu investieren, die im Rahmen dieser Arbeit nicht zurVerfügung stand.
Tabelle 3.1: Synthese von CrOCl aus CrCl3 und TiO2
Vers.
Nr. |
Einwaage (mg) |
Ampullenlänge (cm)
Durchmesser |
Ofenheizung |
Fallen |
Hochvakuum |
| TiO2 |
CrCl3 |
23 mm |
5 mm |
Temperatur
(°C) |
Dauer
(min) |
| 1.1 |
163,0 |
723,0 |
18 |
10 |
400
500
600
700
800
900 |
60
30
30
30
30
30 |
LN2 |
laufende Pumpe
p = 10-4 Torr |
| 1.2 |
159,8 |
633,4 |
18 |
10 |
bis 900 |
60 |
-30
LN2 |
laufende Pumpe |
| 1.3 |
159,8 |
633,4 |
18 |
10 |
900 |
120 |
-40 |
vor dem Heizen evakuiert, danach von der Pumpe getrennt |
| 1.4 |
40,1 |
157,9 |
6 |
15 |
920 |
600 |
--- |
an der Pumpue evakuiert, abgeschmolzen |
| 1.5 |
10,0 |
39,5 |
-- |
10 |
bis 920 |
400 |
Versuch wurde am Massenspektrometer durchgeführt |
| Cr2O3 |
+ |
CrCl3 |
--> |
3 CrOCl |
| 152,0 |
|
158,4 |
|
103,5 |
Da diese Umsetzung bei den notwendigen Temperaturen um 700...1080°C eine Gleichgewichtsreaktion darstellt, ist bei vorgegebenen Ampullengrößen die Ausbeute eine Frage der Ausgangsprodukte und der Temperaturwahl.
Mit Hilfe der bei H. Schäfer(1) angegebenen thermodynamischen Daten wurde versucht, die Parameter der Reaktion soweit zu optimieren, daß das gewünschte Produkt in guter Ausbeute und frei von Verunreinigungen von CrCl3 und Cr2O3 war (Überlegungen zum Transportmechanismus loc. cit. 1, 33).
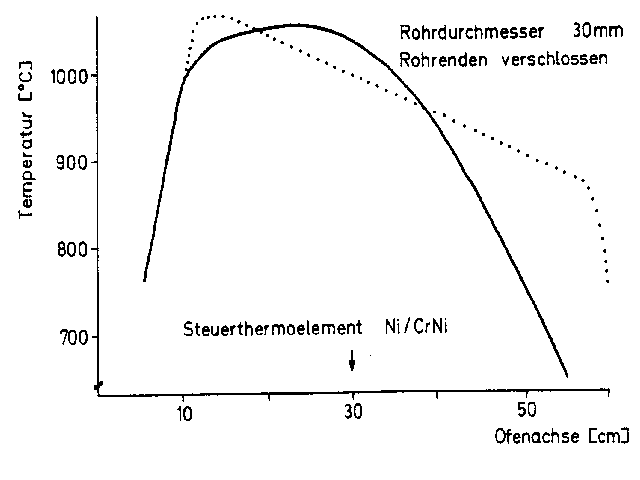
Als erstes Problem stellte sich die Ofenkurve der zur Verfügung stehenden Rohröfen (z.B. Abb 3.3) heraus. Der Bau eines geeigneten Ofens (Temperaturverteilung z.B. Abb 3.3) scheiterte an der mit unseren Mitteln technisch nicht erreichbaren Leistungsdichte an den Enden des Ofens.
Da sich im nicht exakt einstellbaren Temperaturgefälle neben CrOCl auch CrCl3 aus der Gasphase abschied, wurden durch eine Versuchsreihe die Bedingungen für die Reinigung und Trennung durch Sublimation des überschüssigen CrCl3 ohne Zersetzung des CrOCl (damit wird eine Verunreinigung durch Cr2O3 erzeugt) erarbeitet.
Zur Ausbeuteerhöhung wurde in späteren Versuchen ein Cr2O3 eingesetzt, das durch definierte Zersetzung von Ammoniumdichromat erhalten wurde.
Die Bedingungen für die Zersetzung wurden durch thermogravimetrische Untersuchungen bestimmt.
Die Reaktion von CrCl3 mit käuflichem geglühtem Cr2O3 (Merck 2483 rein) ergaben meist nur eine Ausbeute von ca. 6%.
Durch die Verwendung eines oberflächenreicheren Oxids, das durch thermische Zersetzung aus Ammoniumdichromat erhalten wurde, konnte die Ausbeute um ca. 1...4% erhöht werden.
Um eine möglichst niedrige Zersetzungstemperatur zu finden, wurde dieAbspaltung von Wasser und Stickstoff thermographimetrisch verfolgt.
(NH4)2Cr2O7 --> Cr2O3 x H2O + 3 H2O g + N2g
Cr2O3 x H2O --> Cr2O3 + H2O g
Da der erste Abbauschritt der Zersetzung leicht unter Verpuffung stattfindet, mußte das Dichromat mit frisch geglühtem Al2O3 'verdünnt werden.
| Einwaagen: |
8,027 mg |
(NH4)2Cr2O7 |
(Merck 1121 reinst) |
| 61,74 mg |
Al2O3 |
|
Der Gewichtsverlust bis ca. 230°C ist der Abspaltung oberflächlich adsorbierten Wassers zuzuschreiben.
Zwischen 220 und 330°C erfolgt die Umwandlung zu Cr2O3 x H2O unter Abgabe von Wasser und Stickstoff.
Das verbleibende Kristallwasser ist bei ca. 750°C vollständig abgespalten.
Der weitere Anstieg der Abbaukurve oberhalb von 750°C ist auf einen elektronischen Trift des Meßsystems zurückzuführen.
Die Zersetzung läßt sich anhand der Farbänderung des Produkts leicht verfolgen:
Nach der Umwandlung bis ca. 330°C erscheint das Produkt schwarz, bei weiterer Temperaturerhöhung verändert sich die Farbe von schwarz (330°C) über blaugrün (650...900°C) zu hellem gelbgrün (ab ca. 1000°C).
Diffuse Reflexionsspektren von verschieden gefärbten Proben zeigten, daß die Färbungen blaugrün bis hellgrün nur eine Folge des unterschiedlichen Zerteilungsgrades sind, während die schwarze Probe einer anderen Verbindung zuzuschreiben ist.
 Abb 3.4
Abb 3.4: Thermogravimetrie (NH
4)
2Cr
2O
7
Die zu Syntheseversuchen eingesetzten Präparate wurden durch langsames Erhitzen von Ammoniumdichromat bis 750°C im Hochvakuum (p = 10-5 Torr) dargestellt.
Alle Syntheseversuche wurden in Rohröfen von 60 cm Länge (Versuch 2.7: zwei Öfen á 30 cm) in Quarzampullen von 25 cm Durchmesser durchgeführt.
Die Ausgangssubstanzen Chrom(III)-chlorid (Merck 27094 pa wasserfrei) und Chrom(III)-oxid (Merck 2483 rein oder aus (NH4)2Cr2O7) wurden fein zerrieben, unter einem Druck von 9 to/cm² zu Presslingen mit einem Durchmesser von 6 mm geformt.
Die Ampulle wurde nach dem Ausheizen an der Hochvakuumpumpe und dem Beschicken mit den Presslingen auf p = 10-5 Torr evakuiert und abgeschmolzen.
Der Ofen war vor jedem Versuch bereits auf die angegebene Temperatur gebracht und im thermischen Gleichgewicht.
Im ersten Schritt der Synthese reagiert in der heißen Zone CrCl3 mit Cr2O3 über verschiedene Zwischenstufen (vergl. hierzu loc. cit. 1, 33) zu CrOCl, das sich an der kälteren Seite der Ampulle mit nicht reagiertem CrCl3 niederschlägt.
Um das CrCl3 vom gewünschten Produkt zu entfernen, wurde die Ampulle bei der angegebenen Temperatur so lange in den Ofen gelegt, daß das CrOCl an der heißeren Seite im Ofen lag.
In mehreren Versuchen (siehe Versuch 2.7) wurden die Bedingungen für den Transport erarbeitet.
Die Daten und experimentellen Details dieser Syntheseversuche sind in Tabelle 3.2 zusammengefaßt.
Obwohl alle thermodynamischen Angaben bei Raumtemperatur zur Bestimmung der Bedingungen für die Synthese erarbeitet wurden(1), bereitete es große Schwierigkeiten, optimale Ergebnisse zu erhalten.
Große Probleme ergaben die Temperaturwahl und Dauer für die verschiedenen Schritte, ebenso wie die Temperatureinstellung im Ofen.
Durch Variieren der Wickelungsdichte ließ sich die für kommerzielle Rohröfen typische Temperaturverteilung (Abb 3.3) nur wenig verändern, da mit den üblichen Widerstandsdrähten bestimmte Grenzwerte für die Leistungsdichte nicht überschritten werden konnten.
Es gelang zwar, die Temperaturkurve günstig zu beeinflussen, jedoch hielten die Öfen wegen der damit verbundenen Überhitzung der Heizdrähte nur wenige Stunden.
Da aber die beim ersten Syntheseschritt um 825°C liegende Temperatur bereits im steileren Verlauf der Verteilungskurve lag, die exakte Einstellung der Temperatur wegen der sehr nahe beieinanderliegenden Abscheidungstemperaturen von CrCl3 und CrOCl von synthetischer Bedeutung war, wurden Versuche zur Bestimmung dieser Parameter durchgeführt.
Die Versuche im einzelnen (vergl. Tab 3.2):
| Versuch 2.1 und 2.2 |
Beide Synthesen ergaben nur eine Ausbeute von 5,2 bzw. 5,4%.
Das Produkt war (hauptsächlich bei 2.1) stark mit CrCl3 (ca. 7%) verunreinigt;
die ca. 0,5 mm großen schwarzen Blättchen konnten aber unter der Stereolupe von Verunreinigungen gut getrennt werden.
In beiden Fällen schien die kühlere Zone im zweiten Syntheseschritt für eine vollständige Entfernung von CrCl3 zu heiß gewesen zu sein.
Das Präparat aus Versuch 2.2 wurde für den ersten Teil der elektrochemischen Untersuchungen verwendet.
Die bei einer Röntgenaufnahme nach Guinier erhaltenen Linienlagen sind in Tab. 5.2 (Anhang) wiedergegeben und zeigten gute Übereinstimmung mit der Literatur(28).
|
| Versuch 2.3 |
Zur Erhöhung der Ausbeute wurde ein Ansatz fünf Tage im Ofen belassen.
Beim Versuch, die Ampulle aus dem Ofen zu nehmen, kam es zur Explosion.
Die Untersuchung des auffindbaren Ampulleninhalts ergab einen überraschend hohen Anteil an SiO2 auf nicht reagiertem Cr2O3.
Offensichtlich wurde bei derartig langen Reaktionszeiten das Quarzglas schon stark angegriffen.
Auch J. Choy beobachtete ein häufiges zerplatzen der Ampullen nach sehr langen Reaktionszeiten.
Eine genauere Untersuchung der Produkte war nicht mehr möglich.
|
| Versuch 2.4 |
Um die Geschwindigkeit der Sublimation von CrCl3 aus dem Pressling zu verlangsamen, wurde die Reaktionstemperatur im ersten Syntheseschritt bei insgesamt 64-stündiger Reaktionszeit erniedrigt und versucht, das Temperaturprofil etwas flacher zu gestalten.
Die Dauer des zweiten Schrittes wurde aus den bei Versuch 2.1 und 2.2 angegebenen Gründen verlängert.
Diese Maßnahmen führten zueiner Ausbeute von 6,2% schwach und 6,8% stark mit CrCl3 verunreinigten Produkts.
Das nicht reagierte Cr2O3 war schwach mit SiO2 überzogen.
Das Produkt war schichtenweise mit CrCl3 durchsetzt (Temperatursprung im ersten Schritt).
Das schwach verunreinigte ausgelesene Präparat wurde im zweiten Teil der elektrochemischen Untersuchungen eingesetzt.
Ein unter gleichen Bedingungen durchgeführter Parallelversuch mit nicht gepreßten Ausgangssubstanzen ergab eine Ausbeute von nur 6,1% eines mit CrCl3 verunreinigten Präparats.
|
| Versuch 2.5 |
Durch die Erniedrigung des CrCl3-Partialdrucks im ersten und Verlängerung der Behandlungszeitim zweiten Schritt war es gelungen, CrOCl von allem anhaftendem CrCl3 zu befreien.
Allerdings hatte sich dabei bereits ein teil des CrOCl zersetzt und wurde durch einen feinen Überzug von Cr2O3 grün verfärbt.
Die Ausbeute an mechanisch von Cr2O3 befreitem CrOCl betrug 11,9%.
|
| Versuch 2.6 |
In den folgenden Versuchen wurde das aus (NH4)2Cr2O7 erhaltene Oxid verwendet.
Der Einfluß auf die Ausbeute war aber gering,da, wie ein Vergleich mit den Ergebnissen aus Versuch 2.1 und 2.2 zeigt, ebenfalls nur 7,5% erhalten wurden.
Beim Beschicken der Ampulle wurde die gesamte innere Glaswand mit einer feinen Schicht von Cr2O3 bedeckt.
Nach der Reaktion war der mittlere Teil der Ampullenwand frei von Cr2O3, sodaß angenommen werden konnte, daß in diesem Bereich die Reaktionstemperatur optimal gewesen wäre.
|
| Versuch 2.7 |
Um das herausnehmen der Ampulle nach dem ersten Schritt (wobei sich das CrCl3 aus der Gasphase abschied und auch vom Produkt nicht fernzuhalten war) zu vermeiden, wurden zwei Öfen so temperiert, daß die Ampulle zwischen erstem und zweitem Syntheseschritt nur aus einem Temperaturbereich in den zweiten geschoben werden mußte (Abb 3.5).
Das Produkt konnte in zwei Fraktionen unterteilt werden:
- große, mit sehr wenig Cr2O3 verunreinigte Kristalle (4,4%)
- mit wenig CrCl3 verunreinigte Kristalle, die zusammengebacken waren (6,3%)
|
Abb 3.5: Ofenkurve zum Versuch 2.7 (siehe Text)
Tabelle 3.2: Synthesen von CrOCl aus CrCl3 und Cr2O3
Vers.
Nr. |
Einwaage (mg) |
Ampullenlänge (cm)
Durchmesser 25 cm |
Heizung 1. Schritt |
Heizung 2. Schritt |
|
| Cr2O3 |
CrCl3 |
Temperatur
(°C) |
Dauer
(Std) |
Temperatur
(°C) |
Dauer
(Std) |
| 2.1 |
1550 |
1850 |
25 |
1060 / 830 |
24 |
700 / 400 |
10 |
|
| 2.2 |
1310 |
1510 |
24 |
1060 / 820 |
24 |
600 / 400 |
24 |
|
| 2.3 |
1380 |
1585 |
24 |
1060 / 820 |
120 |
--- |
-- |
|
| 2.4 |
1160 |
1210 |
26 |
1050 / 860
1015 / 815 |
40
24 |
700 / 528 |
24 |
|
| 2.5 |
1160 |
968 |
24 |
1057 / 807 |
30 |
700 / 510 |
80 |
|
| 2.6 |
1070 |
1041 |
26 |
1057 / 815 |
24 |
680 / 520 |
20 |
aktiviertes Oxid |
| 2.7 |
948 |
987 |
26 |
1030 / 840 |
48 |
750 / 100 |
3 |
aktiviertes Oxid |
In einer kleinen Versuchsreihe wurden Bedingungen gesucht, CrOCl ohne Zersetzung von anhaftendem CrCl3 zu befreien.
Zusammenfassend ließ sich feststellen, daß bei ca. 530°C / 10-5 Torr die Sublimation von CrCl3 begint.
Hohe Drücke und/oder lange Heizzeiten förderten eine Zersetzung.
In einem Quarzrohr (Durchmesser innen 7 mm) wurde bei ca. 700°C / 10-5 Torr eine Trennung erfolgreich durchgeführt.
Dabei schied sich das CrCl3 in verschiedenen Zerteilungsgraden und nicht näher untersuchten Zersetzungsprodukten als gut getrennte Fraktionen im Rohr ab.
Theorie und Praxis dieser Methode wurden vielfach beschrieben(34, 35) und Untersuchungen dieser Art haben ihren festen Platz bei der Aufklärung von Elektrodenvorgängen.
Im Gegensatz zur kinetischen Polarographie wird bei den hier nicht gerührten Lösungen kein statiionärer Diffusionsstrom erreicht.
Das Vordringen der Diffusionsschicht und daraus resultierende Diffusionsströme können unter Vernachlässigung von Konvektionen rechnerisch erfaßt werden(34, 35, 36).
Ein Ansatz für reversible Reaktionsführung, lösliche Produkte und lineare Diffusion liefert den Peakstrom ip:
| ip = k n3/2 A D1/2 C V1/2 |
|
(34, 35) |
| für das Peakpotential Ep bei der Reduktion: |
| Ep = E1/2 -0,028/n |
|
(34, 37) |
| bzw. Oxidation: |
| Ep = E1/2 +0,028/n |
|
(34, 37) |
| wobei |
ip |
= |
Peakstrom |
[Å] |
| k |
= |
Randles-Sevcik-Konstante
für Cyclovoltametrie beobachtet 2,72E-5 |
(38) |
| n |
= |
Zahl der umgesetzten Elektronen |
[1/mol] |
| A |
= |
Fläche der Arbeitselektrode |
[cm²] |
| D |
= |
Diffusionskoeffizient |
[cm²/s] |
| C |
= |
Konzentration des elektroaktiven Stoffes |
[mol/cm³] |
| V |
= |
Potentialänderung |
[V/s] |
| E1/2 |
= |
Halbstufenpotential |
[V] |
Da jedoch oft (und auch hier bei CrOCl) die Voraussetzungen für obigen Ansatz nur teilweise erfüllt sind, erlaubt diese Methode keine quantitativen oder kinetischen Aussagen;
viele Eigenschaften und Reaktionen einer Elektrode lassen sich jedoch auf einfache Weise erkennen und erklären:
Wichtigste Informationsquelle ist die Knickpunktmethode;
sie liefert das 'Zersetzungspotential Ez' (39).
Obwohl nicht exakt definiert, kann aus ihm dennoch eine Vielzahl von Daten über die elektrochemische Stabilität einer Elektrolytlösung oder das Potential, bei dem das Elektrodenmaterial reagiert, entnommen werden.
Zahlreiche weitere Interpretations- und Diskussionsmöglichkeiten sind bei loc. cit. 40 beschrieben.
Das Blockschaltbild des Versuchsaufbaus ist in Abb 4.1.1 wiedergegeben.
Abb 4.1.1: Blockdiagramm Cyclovoltammetrie
Der Potentiostat als empfindlichste, volltransistorierte Komponente hat folgende Funktion
Auf der (meist geerdeten) Arbeitselektrode wird mit Hilfe der Gegenelektroden ein Potential erzeugt, das bezüglich einer Referenzelektrode ebenso groß ist wie die vorgegebene Sollspannung DU.
Dabei können bei sehr hohen Zellwiderständen an der Gegenelektrode Spannungen bis zu 120 Volt auftreten.
Eine dem fließenden Strom proportionale Spannung und die tatsächliche Spannung zwischen Arbeits- und Bezugselektrode kann belastet (!) abgegriffen und zur Aufzeichnung von Strom-Spannungs-Kurven an einen XY-Schreiber ausgegeben werden.
Für Cyclovoltagramme wird die Sollspannung durch einen Funktionsgenerator in einem vorgegebenen Bereich meist mit 10 mV/s variiert.
Der Strom durch die Elektrode (d.h. die Stromdichte bei bekannter Fläche) wird als Funktion des Potentials registriert.
Besondere Sorgfalt sollte der Erdung des Versuchsaufbaus gewidmet werden.
Verbrummte Cyclovoltagramme sind meist die Ursache unbeabsichtigter Ringerden.
Ein brummfreier Aufbau wird erleichtert durch eine geerdete Arbeitselektrode und erdfreie Ein- bzw. Ausgänge an den Meßgeräten.
Verwendete Geräte:
| Potentiostat |
Wenking |
Modell LB75H |
| Dreieckspannungsgenerator |
Bank |
Modell VSG72 |
| XY-Schreiber |
Rohde & Schwarz |
Modell ZKS 2/.06 |
Den Aufbau der verwendeten Meßzellen gibt Abb 4.1.2 wieder.
Die Fritten, die die Elektrodenräume voneinander trennen, waren bei Cyclovoltagrammen und galvanosatischenExperimenten von der Porengröße D4.
Um die Messungen möglichst reproduzierbar zu machen wurde eine symmetrische Anordnung der Gegenelektroden bezüglich der Arbeitselektrode gewählt.
Die Zellen erlaubten das Arbeiten unter Schutzgas.
Die Bezugselektrode wurde so eingesetzt, daß sie in einer Ebene mit der Probe lag und damit bezüglich der Gegenelektroden den gleichen Abstand hatte wie die Probe.
Die Meßzellen wurden nach jedem gebrauch mindestens 24 Stunden bei 250°C im Trocken schrank ausgeheizt, um die Feuchtigkeit aus den Fritten zu entfernen.
Abb 4.1.2: Meßzelle und mittlerer Elektrodenraum
Das Blockdiagramm des versuchsaufbaus ist in Abb 4.1.3 dargestellt.
Der Potentiostat arbeitet hier als Galvanostat, da er durch die Spannung an den Gegenelektroden den Strom gerade so einstellt, daß der Spannungsabfall an einem zur Meßzelle in Serie geschalteten Widerstand R der vorgegebenen Sollspannung entspricht.
Die Stromrichtung und Stromstärke kann durch die Sollspannung (Polarität, Größe) und R bestimmt werden.
Als Sollspannungsquelle kann eine hinreichend konstante Quelle Verwendung finden; die Belastung ist zu vernachlässigen.
Als Widerstand R wurde ein 10-Gang-Wendel-Potentiometer 100 kOhm verwendet.
Um den Fortgang der Reaktion verfolgen zu können, wurde die Spannung zwischen der Arbeitselektrode und einer Referenzelektrode gegen die Zeit aufgetragen.
Damit das Ruhepotential der Bezugselektrode wegen eines zugeringen Eingangswiderstandes des Schreibers nicht verändert wurde, wurde ein Meßverstärker (Eingangswiderstand 5E12 Ohm) mit einem Verstärkungsfaktor 1 zwischengeschaltet.
Verwendete Geräte:
| Potentiostat |
Wenking |
Modell LB75H |
| Sollspannungsquelle |
Bank |
Modell VSG72 |
| Meßverstärker |
Knick |
Diomod 72W |
| Schreiber |
JJ Instruments |
CR600 Recorder |
Abb 4.1.3: Blockdiagramm Galvanostatik
|
| Tabelle 5.1: |
CrOCl, dargestellt aus CrCl3 und Cr2O3
Guinier-Aufnahme aus Versuch 2.1 |
| d [Å] |
I |
hkl |
|
d [Å] |
I |
hkl |
| 3,4527 |
100 |
101 |
|
1,5558 |
60 |
021 |
| 2,4505 |
100 |
110 / 012 |
|
1,5168 |
80 |
212 |
| 2,3365 |
80 |
111 |
|
1,4429 |
80 |
121 |
| 1,9309 |
80 |
200 |
|
1,2268 |
60 |
220 / 024 |
| 1,8732 |
60 |
201 |
|
1,0212 |
40 |
032 |
| 1,5881 |
80 |
0201 |
|
1,0125 |
40 |
216 |
|
| Tabelle 5.2: |
CrOCl, dargestellt aus CrCl3 und Cr2O3
Guinier-Aufnahme aus Versuch 2.2 |
| d [Å] |
I |
hkl |
|
d [Å] |
I |
hkl |
| 3,4510 |
100 |
101 |
|
1,9990 |
60 |
013 |
| 2,4535 |
100 |
110 / 012 |
|
1,9320 |
80 |
200 |
| 2,3391 |
80 |
111 |
|
1,8727 |
60 |
201 |
| 2,1336 |
30 |
103 |
|
1,5889 |
80 |
020 |
| 2,0732 |
30 |
112 |
|
1,5553 |
60 |
021 |
| 1. |
H. Schäfer, F. Warthenpfuhl:
Z. anorg. Chem., 308, 282 (1961) |
| 2. |
S. K. Deb, R. F. Shaw:
US. Patent 3 521 941 (1970) |
| 3. |
S. K. Deb:
Applied Optics Suppl., 192 (1969) |
| 4. |
P. G. Dickens, M. S. Whittingham:
Quart. Rev. Chem. Soc., 22, 30 (1968) |
| 5. |
B. T. Matthias, M. Marezio, E. Cornzwit, A. S. Cooper, H. E. Barz:
Science, 175, 1465 (1972) |
| 6. |
M. Sudo, M. Ichikawa, M. Soma, T. Onishi, K. Tamaru:
J. Phys. Chem., 73, 1174 (1969) |
| 7. |
V. I. Mashinski, V. A. Postnikov, Yu. N. Novikov, A. L. Lapidus, M. E. Vol'pin, Ya. T. Eidus:
Izv. Akad. Nauk SSSR, Ser. Khim., 9, 2018 (1976) |
| 8. |
M. A. Boersma:
Catal. Rev., 10, 243 (1974) |
| 9. |
Structure Reports, 27, 469 |
| 10. |
Jin-ho Choy:
Dissertation, Ludwig-Maximilians-Universität, München |
| 11. |
M. S. Whittingham in:
Intercalated Layered Materials, ed.
F. Levy, D. Reidel Publishing Comp. Dordrecht Holl. (1979) |
| 12. |
M. S.. Whittingham:
J. Electrochem. Soc., 123, 315 (1976) |
| 13. |
M. S. Whittingham:
Prog. in Solid State Chem., 12, 41 (1978) |
| 14. |
B. G. Silbernagel:
Solid State Commun., 17, 361 (1975) |
| 15. |
M. S. Whittingham in:
Electrode Materials and Processes for Energy Conversion and Storage, ed.
J. D. E. McIntyre, S. Srinivason, F. G. Will
The Electrochemical Society (1977) |
| 16. |
M. S. Whittingham:
Prog. in Solid State Chem., 12, 41 (1978) |
| 17. |
R. Schöllhorn, H. Meyer:
Mat. Res. Bull., 9, 1237 (1974) |
| 18. |
M. S. Whittingham:
US. Patent 4 009 052 |
| 19. |
D. W. Murphy, J. N. Carides, F. J. DiSalvo, C. Cros, J. Waszcak:
Mat. Res. Bull., 12, 825 (1977) |
| 20. |
Gmelin:
Handbuch der Anorganischen Chemie
No. 52 A.2 Chrom
Verlag Chemie Weinheim/Bergstraße 1963 |
| 21. |
J. T. Kummer:
Prog. in Solid State Chem., 7, 141 (1972) |
| 22. |
H.. Meyer, A. Weiss, J. O. Besenhard:
Mat. Res. Bull., 13, 913 (1978) |
| 23. |
J. P. Venién, P. Palvadeau, D. Schleich, J. Rouxel:
Mat. Res. Bull., 14, 891 (1979) |
| 24. |
A. H. Thompson, M. S. Whittingham:
Mat. Res. Bull., 12, 741 (1977) |
| 25. |
M. S. Whittingham:
US Patent 4 049 887 |
| 26. |
M. Armand, L. Coic, P. Palvadeau, J. Rouxel:
J. Power Sources, 3, 137 (1978) |
| 27. |
J. Dahn, R. R. Haering:
Mat. Res. Bull., 14, 1259 (1979) |
| 28. |
A. N. Christensen, T. Johansson, S. Quézel:
Acta Chem. Scand., A28, 1171 (1975) |
| 29. |
G. W. Watt, U. Kask:
J. Inorg. Nucl. Chem., 27, 1925 (1925) |
| 30. |
D. Kiraly, K. Zalatnai, M. T. Beck:
J. Inorg. Nucl. Chem., 11, 170 (1959) |
| 31. |
W. Frank:
US Patent 3 134 640 (1964) |
| 32. |
Ch. Steinle:
Zulassungsarbeit Ludwig-Maximilian-Universität München
Prof. Boehm (1975) |
| 33. |
G. Mair:
Bericht zum anorg. Fortgeschrittenenpraktikum
Ludwig-Maximilian-Universität München
Prof. A. Weiss (1980) |
| 34. |
P. Delahay:
New Instrumental Methods in Electrochemistry
Interscience Publishers, New York 1954 |
| 35. |
R. N. Adams:
Electrochemistry at Solid Electrodes
Marcel Dekker, New York (1969) |
| 36. |
J. E. B. Randles:
Trans. Faraday Soc., 481, 327 (1948) |
| 37. |
H. Matsuda, Y. Ayabe:
Z. Elektrochem., 59, 494 (1955) |
| 38. |
T. R. Mueller, R. N. Adams:
Anal. Chim. Acta, 25, 482 (1961) |
| 39. |
R. Brdicka:
Grundlagen der physikalischen Chemie
VEB Deutscher Verlag der Wissenschaften
Berlin (1968), S. 749 |
| 40. |
M. Shaw, A. H. Remanick:
Electrochemical Characterisation of Systems for Secondary Battery Application
NASA-Reports N66-35664 / NASA-CR-62034 (1966) |
| 41. |
M. M. Baizer:
Organic Electrochemistry
Marcel Dekker, New York (1973) |
| 42. |
W. R. Turner, P. J. Elving:
Anal. Chem., 37, 467 (1965) |