
| MENU: |
|---|
| ASSUNTOS M�DICOS GERAIS: |
|---|
| ASSUNTOS PARA OS PROFISSIONAIS DAS �REAS DE SA�DE E AFINS: |
|---|
Desenvolveremos esse tópico seguindo a seqüência apresentada no Quadro 1, e baseados no trabalho de Triplett e no de Rao e Gabbeta, com pequena modificação.
Ressaltamos que a vWD foi incluída porque, apesar de ser um distúrbio plasmático, altera a função plaquetária. Entretanto, o comentário sobre ela já foi feito em Distúrbios Hereditários da Coagulação. Também a afibrinogenemia congênita, apesar de pertencer aos distúrbios plasmáticos hereditários, foi citada nesse grupo por ser responsável por alteração da agregação plaquetária.
|
Anormalidades relacionadas à adesividade plaquetária: Doença de von Willebrand Pseudo von Willebrand (tipo plaquetário semelhante à vWD) Síndrome de Bernard Soulier (anomalia ou ausência da GP Ib) Anormalidade da agregação plaquetária: Afibrinogenemia congênita Trombastenia de Glanzmann (anormalidade da GP IIb/IIIa) Deficiência de substâncias do "pool" de armazenamento: Deficiência dos corpos densos: Síndrome de Hermansky-Pudlak Síndrome de Chediak-Higashi Trombocitopenia com ausência de rádio Deficiência dos grânulos alfa: Síndrome da plaqueta cinza Defeitos dos sinais de transdução: Anormalidade da via do ácido araquidônico (AA): Diminuição da liberação do AA Deficiência da ciclo-oxigenase Defeitos na interação plaqueta-agonistas: Defeitos dos receptores: TxA2, colágeno, ADP, epinefrina Defeitos na ativação da proteína G: Deficiência de G (alfa)q Defeitos no metabolismo de fosfoinositide: Deficiência de PLC-ß 2 Defeitos na mobilização de cálcio Defeitos na fosforilação de proteína Defeitos na regulação do cito-esqueleto: Síndrome de Wiskott-Aldrich Desordem da interação de fatores da coagulação com a membrana plaquetária: Síndrome de Scott |
Quadro 1: Desordens funcionais plaquetárias baseadas no trabalho de Triplett e no de Rao e Gabbeta, com pequena modificação.
Esse grupo de distúrbios tem como manifestação clínica hemorragias de diversas intensidades, freqüentemente mucocutâneas ou após cirurgias e traumas.
As desordens plaquetárias congênitas não podem ser, freqüentemente distinguidas com bases, apenas, nas manifestações clínicas. Como já nos referimos anteriormente, o estudo laboratorial é destaque de importância diagnóstica. Entretanto, para chegar ao esclarecimento de muitos distúrbios nesse grupo, são imprescindíveis exames sofisticados, por meio de instrumentos ou de exames bioquímicos que, somente, são efetuados em laboratórios de coagulação altamente especializados.
O tempo de sangramento aumentado é freqüentemente visto nesses distúrbios, e no exame do esfregaço de sangue observam-se freqüentes plaquetas gigantes.
O estudo da agregação plaquetária, por meio do agregômetro, com o uso de diversos agonistas (ADP, colágeno, epinefrina e ristocetina) é importante para o diagnóstico desse grupo de distúrbios.
Na Figura 1 apresentamos um desenho, esquemático, das principais reações desencadeadas pelos indutores fisiológicos da agregação plaquetária, para podermos compreender melhor os distúrbios relacionados à transdução de sinal.
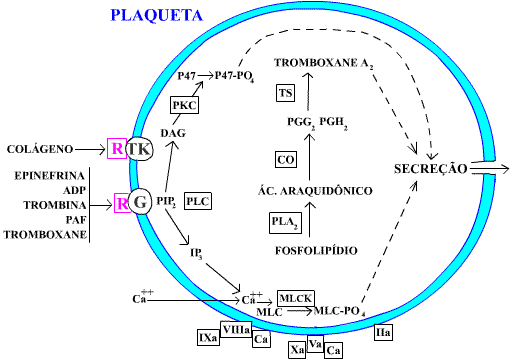
Figura 1: desenho esquemático das reações plaquetárias que ocorrem após o estímulo dos indutores de agregação.
CO = ciclo-oxigenase, PKC = proteinoquinase, G = proteínas G, MLC = cadeia leve da miosina, MLCK = MLCquinase, R = receptor, DAG = diacilglicerol, TK = tirosinoquinase, TS = tromboxane-sintetase, PAF = fator ativador de plaqueta,
IP3 = trifosfato de inositol, PLA2 = fosfolipase A2, PIP2 = bifosfato de fosfatidil-
inositol.
O desenho da Figura 1 é baseado no de Rao e Gabbeta, porém, simplificado. Tivemos a
intenção de destacar, apenas, as reações que representam a transdução dos sinais que chegam à
membrana plaquetária, e que são responsáveis pela indução da secreção de diversas substâncias
plaquetárias.
Como em outras células, a ativação plaquetária e a secreção, são reguladas pelas alterações ao
nível de ATP, influxo de cálcio, hidrólise dos fosfolipídios da membrana da plaqueta e
fosforilação de diversas proteínas intracelulares.
Como é mostrado na Figura 1, essas reações têm início quando o efetor se liga a um receptor
específico que, por sua vez, interage com a proteína G. As proteínas G constituem um grupo
heterogêneo de proteínas, encravadas na membrana, que têm a propriedade de ligar os receptores
com as enzimas efetoras intracelulares.
A partir da interação, específica, do efetor com seu receptor e a proteína G, dá-se a hidrólise
do fosfolipídio da membrana plaquetária - fosfatidilinositol 4, 5 - bi-fosfato (PIP
2). Com isso são gerados o diacilglicerol (DAG) e o trifosfato de inositol (IP3). Esse último medeia o movimento de cálcio no citosol plaquetário que, por sua
vez, estimula a fosfolipase A2 (PLA2) na reação que gera
tromboxane A2 e produz a fosforilação da cadeia leve da miosina.
A miosina, ao interagir com a actina, promove, por contração, a compressão dos constituintes do
citosol plaquetário, inclusive dos corpos densos e grânulos alfa, e com isso leva à secreção de
substâncias. A actina e a miosina também promovem a mudança de forma das plaquetas.
A DAG ativa a proteinoquinase C (PKC) que leva à fosforilação de diversos substratos, inclusive
da P47, que contribuem para a secreção plaquetária.
No Quadro 2 apresentaremos as características mais importantes para o esclarecimento
diagnóstico dos distúrbios funcionais plaquetários.
| Pseudo vW (P-vW) | É muito semelhante ao tipo 2B da vWD. Pode apresentar alterações do TS, FVIII-C, vWF(ag) e vW:R CoF. A agregação plaquetária está aumentada com baixas concentrações de ristocetina (0.3-0.5mg/ml). Pode haver discreta trombocitopenia. Ao contrário do tipo vWD 2B onde o defeito está no vWF, aqui o defeito está no receptor plaquetário GP Ib-IX-V para o vWF. Esse defeito torna o referido receptor mais ávido para os grandes multímeros do vWF. Como conseqüência dessa avidez, os grandes multímeros são consumidos. |
| Síndrome de Bernard-Soulier (SB-S) | Freqüentemente autossômica recessiva e associada à consangüinidade. Pacientes heterozigotos apresentam 50% da quantidade normal do receptor plaquetário GP Ib-IX-V. Há discreto sangramento ou até mesmo ausência. Casos de SB-S homozigotos são raríssimos. O tempo de sangramento pode estar aumentado e no esfregaço de sangue são vistas plaquetas gigantes. Pode haver trombocitopenia. A anormalidade mais característica está no teste de agregação plaquetária (RIPA) que é normal para os diversos agonistas fisiológicos (ADP, epinefrina, colágeno) mas somente alterada para a ristocetina. A diferenciação com a vWD está em que a adição de plasma normal não corrige o defeito de agregação na SB-S ao contrario da resposta na vWD. Testes bioquímicos, eletroforese em SDS agarose, auto-radiografia, "imunoblotting" de lisado plaquetário com anticorpo anti-GP Ib-IX-V, confirma o diagnóstico ao determinar a deficiência da referida glicoproteina. |
| Afibrinogenemia congênita | Nesse distúrbio, devido à falta de fibrinogênio para fazer a ligação entre duas plaquetas (entre as GP IIb-IIIa), a curva de agregação plaquetária terá a mesma característica da observada na trombastenia de Glanzmann e que será comentada a seguir. |
| Trombastenia de Glanzmann | A Trombastenia de Glanzmann é uma doença autossômica recessiva que decorre de um distúrbio funcional da GP IIb-IIIa. Esse distúrbio deriva da mutação nos genes que codificam as GP IIb e IIIa, com as conseqüentes alterações quantitativa e qualitativa das referidas glicoproteinas. Manifesta-se clinicamente por sangramento muco-cutâneo, particularmente epistaxe, púrpura e menorragia. A alteração laboratorial, mais característica, é a intensa redução ou a ausência da agregação plaquetária, com o emprego do agregômetro (RIPA), aos diversos agonistas fisiológicos. A curva característica é a de tipo C da Figura 2 do tópico Testes Laboratoriais. |
| Deficiência de substâncias do "pool" de armazenamento |
Em alguns distúrbios desse grupo o defeito está relacionado à diminuição de substâncias armazenadas nos corpos densos e nos grânulos alfa (citados no Quadro 1). Na curva de agregação plaquetária, a segunda parte da curva está ausente (curva B da Figura 2 do tópico Testes Laboratoriais), a qual corresponde à agregação pelas substâncias liberadas na ativação plaquetária. Os distúrbios congênitos desse grupo foram mencionados no Quadro 1. |
| Defeitos dos sinais de transdução | Ao contrário do grupo anterior, nesse grupo, a deficiência de substâncias no pool de armazenamento, decorre de um distúrbio localizado no mecanismo que é desencadeado quando o receptor da membrana plaquetária é ativado pelo agonista. O distúrbio pode acontecer em diversos setores desse mecanismo, desde o momento da ação sobre o receptor até a produção dos efetores responsáveis pela liberação das substâncias do pool de armazenamento, bem como das substâncias formadas em diversas reações (ver Figura 1). Na curva de agregação plaquetária (RIPA), está ausente a segunda onda (estímulo endógeno), o mesmo defeito verificado no grupo anterior. É importante assinalar que o ácido acetil salicílico também interfere na produção dessa segunda parte da curva de agregação plaquetária, ao inibir a ciclo-oxigenase e diminuir a produção do tromboxane A2. O artigo de Rao e Gabbeta trata, especificamente, das desordens congênitas da transdução do sinal que chega à membrana plaquetária. |
| Desordem da interação de fatores da coagulação com a membrana
plaquetária |
A síndrome de Scott é uma desordem hemorrágica rara na qual, as plaquetas e outras células sangüíneas falham em promover a união de fatores plasmáticos (Va e Xa) com a membrana celular. O aumento da quantidade da fosfatidilserina, carregada negativamente, no folheto externo da membrana plaquetária é essencial para a formação da superfície coagulante. Sabe-se que o defeito, na síndrome de Scott, decorre da incapacidade em mobilizar a fosfatidilserina da camada interna da membrana celular para a camada externa, em resposta à elevação do Ca2+ na face interna da membrana (Zhou). |
Quadro 2: Características principais das diversas desordens funcionais plaquetárias.